Resumen
Entrectinib y larotrectinib son dos nuevos fármacos de molécula pequeña activos por vía oral que ejercen una actividad inhibitoria de tirosina cinasas, competitiva con ATP y selectiva para los receptores de la tropomiosina cinasa TRK (TRKA, TRKB y TRKC); entrectinib además inhibe la tirosina cinasa ROS1 y la cinasa del linfoma anaplásico (ALK). Todas ellas son proteínas con potencial tumorigénico que, cuando se encuentran sobreactivadas por mutación en sus genes, determinan la hiperactivación de las vías de señalización descendentes (como las de MAPK o PI3K/AKT) que da lugar a una proliferación celular ilimitada. Han recibido autorización condicional para el tratamiento por vía oral en monoterapia de pacientes adultos y pediátricos con tumores sólidos que presentan una fusión del gen NTRK y tienen enfermedad localmente avanzada, metastática o cuya resección quirúrgica probablemente genere una elevada morbilidad, y con ausencia de opciones terapéuticas satisfactorias.
La eficacia clínica de entrectinib se probó en 3 ensayos clínicos de fase 1 y 2, no controlados y de un solo brazo; con datos de hasta 150 pacientes adultos con tumor avanzado con fusión NTRK+ y localización primaria extracraneal, la mayoría de los cuales se incluyeron en el estudio multicéntrico y de fase 2 STARTRK-2. El fármaco aporta una tasa de respuesta global del 61% (completa en el 17% de los casos), y una mediana de duración de la respuesta de 20 meses, de forma que permanecen con respuesta tras al menos 1 año dos tercios de los pacientes respondedores.
Además, entrectinib ha sido autorizado en cáncer de pulmón no microcítico avanzado ROS1+, con datos de 168 pacientes con tumor recurrente o metastásico no pretratados con inhibidores de ROS, en los que mostró una tasa de respuesta objetiva del 68% (solo 9% completas). En ambas indicaciones, en el perfil de seguridad de entrectinib sobresalen por su frecuencia (> 10%) las siguientes reacciones adversas: alteraciones del tracto gastrointestinal (disgeusia, estreñimiento, diarrea, náuseas y vómitos), fatiga y dolores musculo-articulares, y alteraciones del perfil bioquímico sugerentes de hepatotoxicidad (elevación de creatinina y transaminasas).
Por otro lado, de cara a la evaluación de la eficacia clínica de larotrectinib se dispone de un análisis combinado de 3 ensayos aún en marcha en fases tempranas de la investigación clínica y heterogéneos en diseño (abiertos, multicéntricos y de un solo brazo) que, en conjunto, enrolaron a 313 pacientes adultos y pediátricos con tumores sólidos localmente avanzados o metastásicos de diversa histología y localización. En pacientes con afectación extracraneal primaria se alcanzó una tasa de respuesta global del 67% (23% de respuestas completas) y en quienes tenían tumor primario en el SNC, esa tasa fue del 61% (20% de respuestas completas). Su efecto antitumoral es rápido (mediana de tiempo hasta la respuesta de < 2 meses) y duradero, con una mediana de duración de la respuesta de 43 meses. En el perfil de seguridad de larotrectinib destaca una alta notificación de eventos adversos relacionados con su uso (80%), pero casi todos son leves-moderados en severidad, y ninguno resultó fatal. Las reacciones adversas más frecuentes (≥ 20%) fueron: fatiga, hepatotoxicidad con aumento de transaminasas, mareo, alteraciones digestivas (estreñimiento, náuseas y vómitos) y anemia.
Estamos ante dos medicamentos que han supuesto un cambio de paradigma en el abordaje de determinados tipos tumorales –del enfoque centrado en histología/localización al centrado exclusivamente en el perfil molecular–, pero que requieren de un procedimiento de estudio genético previo que no se realiza de forma rutinaria en la práctica habitual en España. Sea como fuere, el número de pacientes que podrán usar larotrectinib y entrectinib será pequeño, por la baja frecuencia (< 1%) de las fusiones NTRK+ en el conjunto de tumores sólidos.
Aspectos fisiopatológicos
La familia de genes del receptor de tirosina cinasa neurotrófico (NTRK, por sus siglas del inglés Neurotrophic Tyrosine Receptor Kinase) comprenden tres genes –NTRK1, NTRK2 y NTRK31 – codificantes para los receptores de tropomiosina – TRKA, TRKB y TRKC, respectivamente–, unas proteínas transmembrana que se localizan principalmente en tejido neuronal. En condiciones fisiológicas, por unión de alta afinidad a ellos de distintos factores de crecimiento2 (como las neurotrofinas), estos receptores se dimerizan y van a promover la activación de diferentes vías de señalización celular como PI3K, MAPK y JAK-STAT, o PLC1-4, facilitando, entre otras funciones, la proliferación celular y la supervivencia neuronal, jugando un papel clave en el desarrollo y funcionalidad del sistema nervioso central y periférico.
Se han identificado una serie de reordenamientos génicos que determinan la fusión de genes que codifican el dominio tirosina cinasa de los TRK y resultan en la síntesis de proteínas con una activación patológicamente constitutiva del dominio cinasa: en consecuencia, se sobreactivan las vías de señalización implicadas en la proliferación y supervivencia celular, lo que conduce a un crecimiento celular anómalo y a la aparición y desarrollo de tumores.
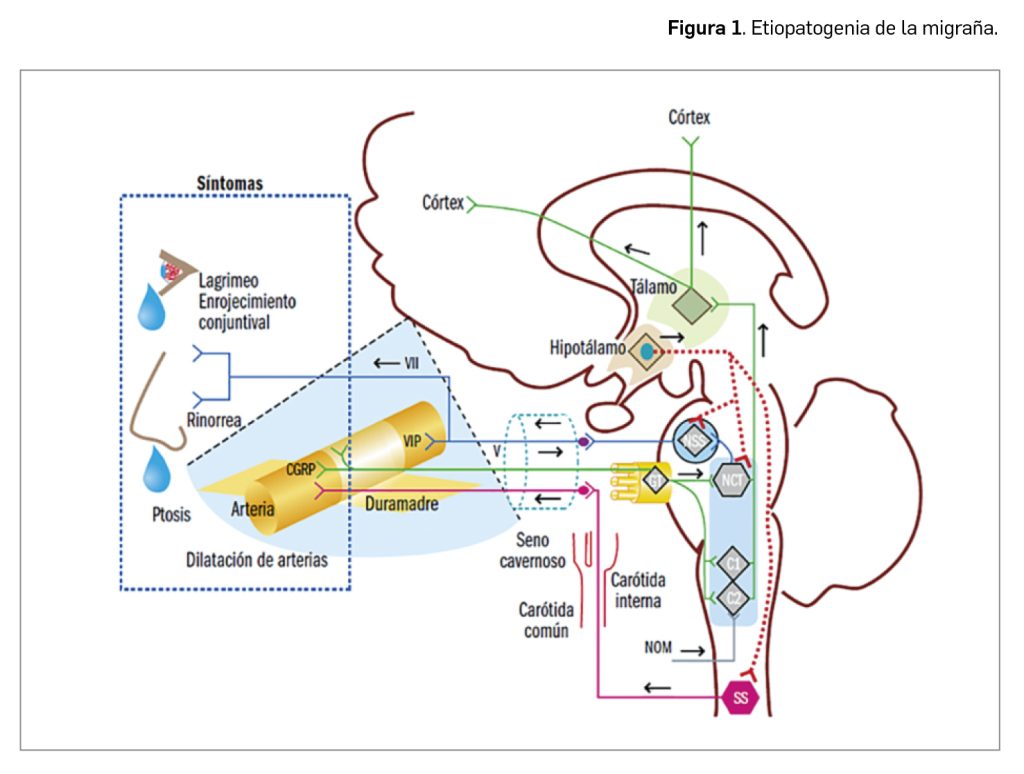
Durante estos reordenamientos el extremo 3´ (C-terminal) del gen NTRK codificante para el dominio tirosina cinasa de la proteína TRK se fusiona con la parte 5´ (N-terminal) del gen acompañante, que usualmente incluye un dominio de dimerización, proceso que se producirá ahora de manera independiente de ligando (Figura 1).
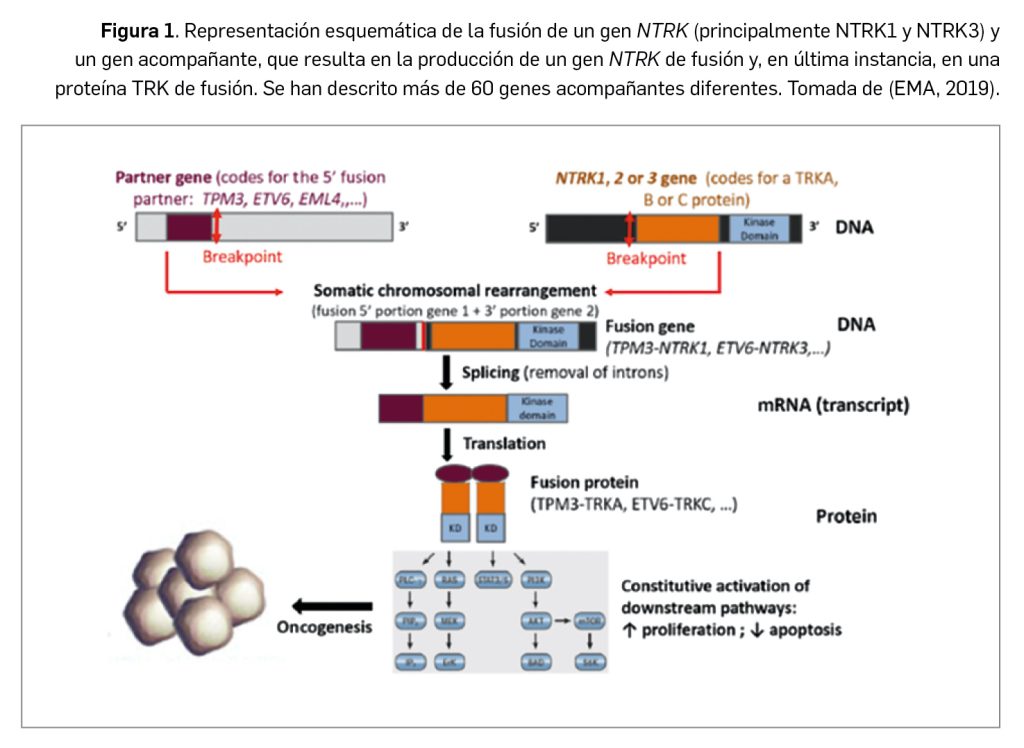
La primera descripción de una fusión de genes NTRK se hizo en 1986, en un tumor colorrectal, y, desde entonces, gracias al progreso de las técnicas de investigación genómica, se ha identificado esta clase de anomalías en distintos tipos de tumores sólidos tanto en adultos como niños, con frecuencias variables, aunque baja de forma global: se considera una anormalidad genética rara que puede encontrarse en aproximadamente el 0,25%-1% del total de tumores sólidos diagnosticados, por debajo siempre del 0,5% de los casos de cánceres más incidentes, como los de colon-recto, mama, pulmón, páncreas o melanoma. En algunos tumores con una prevalencia media se han descrito estos reordenamientos en una mayor proporción de casos, como en colangiocarcinoma (4%), glioblastomas (6%) o tumores papilares de tiroides (30%).
Sin embargo, la identificación de oncogenes NTRK puede disparar su frecuencia hasta más del 90% de algunos tumores sólidos raros, como los siguientes: carcinoma secretor de mama, carcinoma secretor análogo mamario de glándulas salivares3, el fibrosarcoma congénito infantil o el nefroma mesoblástico congénito de tipo celular. E incluso se han visto frecuencias de aparición intermedias (5-25% de los casos) en otros tumores también poco comunes, como el tumor de Spitz, el glioma del tronco encéfalo infantil o los tumores gastrointestinales estromales, entre otros.
Hasta hace poco tiempo, no se ha prestado especial relevancia a estas alteraciones genéticas en las decisiones terapéuticas, en parte por la ausencia de terapias dirigidas. Cada uno de los tumores sólidos se ha tratado clásicamente siguiendo las recomendaciones de consenso según el órgano afectado (en base a histología) y el estadio, en muchos casos con ausencia de opciones terapéuticas satisfactorias4. Grosso modo, los principales tipos de tratamientos oncológicos son cirugía y radioterapia (estos dos aplicables sobre todo en el diagnóstico primario, con finalidad fundamentalmente paliativa en el contexto de un cáncer localmente avanzado o metastásico), así como la quimioterapia citostática/citotóxica, la terapia dirigida, la inmunoterapia o la terapia hormonal como opciones de tratamiento sistémico.
La posibilidad de tratar los tumores con reordenamientos de NTRK, es decir, en base a las alteraciones moleculares y con independencia de su localización o histología (tumor agnóstico), ha emergido de manera interesante con la aparición y aprobación de los fármacos que se comentan en el presente artículo, y gracias a la evolución de las técnicas de diagnóstico molecular. En este sentido, la detección de fusiones NTRK en muestras de tumor puede hacerse a través de técnicas como la fluorescencia con hibridación in situ (FISH, por sus siglas en inglés), la inmunohistoquímica, la reacción en cadena de la polimerasa con transcripción reversa o las técnicas de secuenciación genómica de próxima generación (NGS, por sus siglas en inglés). La aplicación de estas últimas técnicas sobre el material genético del tumor aporta la información más completa, y, aunque de mayor coste y complejidad tecnológica, sería la prueba diagnóstica preferente o gold standard en la detección de fusiones de NTRK (EMA, 2019).
Conviene recordar que, en líneas generales, los pacientes con tumores sólidos avanzados –a nivel local o sistémico– presentan manifestaciones clínicas importantes que comprometen su supervivencia y, desde una perspectiva psicosocial, su enfermedad interfiere sustancialmente en su calidad de vida y la de su familia, siendo frecuente la afectación emocional (miedo, ira, dolor, ansiedad, depresión, soledad, etc.) y el requerimiento de atención psicológica. Estos pacientes representan, pues, una necesidad médica no cubierta, y la individualización de su abordaje persigue maximizar los beneficios y minimizar los daños de las intervenciones terapéuticas.
Por último, se debe hacer una mención a un gen que juega un papel importante en el cáncer de pulmón no microcítico (en adelante, CPNM)5: el protooncogén ROS1. Situado en el cromosoma 6, codifica para un receptor tirosina cinasa huérfano, sin un ligando conocido, cuya función fisiológica aún no está del todo clara, pero para el que se han descrito una serie de translocaciones cromosómicas que pueden resultar en reordenamientos –fusiones– del gen. De forma similar a lo descrito para NTRK, tras una primera identificación en un CPNM en 2007, se han descrito hasta 22 genes acompañantes que pueden participar en la fusión con ROS1 en cáncer de pulmón, siendo la fusión CD74-ROS1 el reordenamiento más común. Tales eventos de fusión conducen a la activación constitutiva de la cinasa ROS1, lo cual impulsa la transformación celular y promueve la supervivencia y la proliferación celular a través de las vías de señalización de SHP-1/SHP-2, JAK/STAT, PI3K/AKT/mTOR y MAPK/ERK.
En las últimas décadas, una serie de alteraciones moleculares identificadas en los CPNM, sobre todo en los tumores avanzados, han modificado sustancialmente su tratamiento por el desarrollo y aprobación de terapias dirigidas frente a las tirosina cinasa específicas (además del progreso con los tratamientos biológicos con inhibidores de puntos de control inmunológico dirigidos a PD-1/PD-L1); en líneas generales, esas mutaciones se observan de forma no superpuesta, si bien entre el 1-3% de tumores pueden llegar a tener alteraciones concurrentes. Así, actualmente se dispone de inhibidores de EGFR (tales como erlotinib, afatinib, gefitinib, osimertinib y dacomitinib), de inhibidores de ALK (crizotinib, ceritinib, alectinib, brigatinib y lorlatinib) y de inhibidores de BRAF V600 (dabrafenib combinado con trametinib), todos ellos susceptibles de alteraciones con potencial oncogénico.
Para centrar los enfoques terapéuticos, se acepta que en los CPNM de tipo no escamoso y estadios avanzados deben realizarse sistemáticamente pruebas moleculares para detectar las citadas alteraciones, pero esa investigación molecular no se recomienda para los pacientes con diagnóstico de tumor escamoso (salvo casos inusuales como en no fumadores o exfumadores prolongados). Frente a aquellos tumores que presenten reordenamientos de ROS1, descritos en aproximadamente el 1-2% de los pacientes con CPNM, está aprobado crizotinib, un inhibidor de múltiples tirosina cinasas (ALK, ROS1 y MET) y único fármaco indicado en la UE para tratar pacientes con CPNM avanzado positivo para ROS1, pese a que están bajo investigación otros inhibidores de ROS1 cinasa, como los fármacos de 1ª generación ceritinib, brigatinib y cabozantinib, o los inhibidores de 2ª generación lorlatinib o repotrectinib.
Los resultados más actualizados de su ensayo pivotal de fase 1/2 con una muestra de 53 pacientes con ese tipo de tumores avanzados (estadio IV), tras un seguimiento de más de 5 años, revelan una tasa de respuesta objetiva del 72%, con un tiempo de tratamiento hasta la respuesta de casi 8 semanas. Además, se vio una mediana de duración de la respuesta de 25 meses, una mediana de supervivencia libre de progresión de 19 meses y una mediana de supervivencia global que supera los 51 meses, de modo que la probabilidad de supervivencia a los 4 años es del 51%.
La Sociedad Europea de Oncología Médica recomienda el uso preferente de crizotinib en monoterapia en primera o segunda línea en pacientes con CPNM ROS1+ en estadio IV. Pero si los pacientes fallan a una primera línea con crizotinib, se considerará una siguiente terapia basada en platino. Y es que el desarrollo de resistencias a crizotinib es un escollo importante en su uso clínico, pues el tumor progresa eventualmente en la gran mayoría de pacientes tratados. Tales resistencias pueden producirse por mutaciones6 en los sitios de unión del fármaco en el propio dominio tirosina cinasa de ROS1 o por otros mecanismos “externos”, que incluyen la activación de vías de señalización de derivación (EGFR, RAS y KIT) y cambios fenotípicos como transición epitelial a mesenquimatosa.
Acción y mecanismo
Entrectinib es un nuevo inhibidor de tirosina cinasa activo por vía oral, competitivo con ATP y selectivo para los receptores de la tropomiosina cinasa TRK (TRKA, TRKB y TRKC), la tirosina cinasa ROS y de la cinasa del linfoma anaplásico (ALK). En base a ello, el medicamento ha sido autorizado en monoterapia para el tratamiento de pacientes adultos y pediátricos de al menos 12 años con tumores sólidos que expresan una fusión del gen NTRK y tienen enfermedad avanzada (localmente avanzada, metastásica o donde es probable que una resección quirúrgica provoque una morbilidad severa), si no han recibido previamente un inhibidor de NTRK ni tienen opciones terapéuticas satisfactorias; también está aprobado para tratar pacientes adultos con CPNM avanzado ROS1-positivo no tratados previamente con inhibidores de ROS.
Las proteínas de fusión que incluyen los dominios de las cinasas TRK, ROS1 o ALK ejercen su potencial tumorigénico mediante la hiperactivación de las vías de señalización descendentes, lo que da lugar a una proliferación celular ilimitada. La actividad inhibitoria de entrectinib sobre ellas supone, precisamente, la inhibición de una serie de rutas bioquímicas –oncogénicas– que incluyen a la fosfolipasa C gamma (PLCγ), la proteína cinasa activada por mitógenos (MAPK) y la fosfoinositol-3-cinasa/proteína cinasa B (PI3K / AKT), lo que se traduce en un bloqueo o reducción de la proliferación celular y la inducción de la apoptosis de las células tumorales, habiéndose demostrado su efecto antitumoral in vitro e in vivo en líneas celulares derivadas de múltiples tipos de cáncer (incluidos subcutáneos e intracraneales) que albergan genes de fusión NTRK, ROS1 y ALK.
Durante su desarrollo clínico se demostró que entrectinib es capaz de atravesar la barrera hematoencefálica y alcanzar niveles terapéuticos en el SNC, dado que se comporta como sustrato débil de la glicoproteína-P. La inhibición que entrectinib ejerce sobre las cinasas diana es potente, situándose los valores de CI50 en el rango nanomolar (0,1 a 2 nM); su principal metabolito activo (M5) también ha mostrado una potencia y espectro de actividad similar en estudios in vitro. Es preciso indicar que se han identificado mutaciones7 que han confieren resistencia al tratamiento con entrectinib, aunque aún se desconoce si estas aparecen de novo o si los tratamientos previos con otros fármacos que inhiben las mismas cinasas pueden conferir resistencia cruzada a entrectinib (EMA, 2020; AEMPS, 2022a).
Por su parte, larotrectinib fue el primer inhibidor del receptor de la tropomiosina cinasa diseñado racionalmente para evitar la actividad sobre otras enzimas. También con un mecanismo competitivo con ATP, su diana es específicamente la familia de proteínas TRK (TRKA, TRKB y TRKC), a las que inhibe con alta potencia (valores de CI50 entre 5 y 12 nM) y selectividad (la única actividad inhibitoria sobre otra cinasa se produce a concentraciones 100 veces más altas). Así, ante proteínas quiméricas de fusión TRK oncogénicas –resultado de reordenamientos en los genes NTRK– que tienen activada constitutivamente su actividad tirosina cinasa, el fármaco es capaz de inhibir esa sobreactivación e impedir el exceso de señalización que se produciría de las vías descendentes implicadas en la proliferación y la supervivencia de las células y el consiguiente desarrollo tumoral.
El medicamento fue el primero autorizado en la UE con una indicación agnóstica, o sea, sin especificar el tipo o localización del tumor, sino que valdrá para tratar cualquier tumor con fusión de genes NTRK. Se ha aprobado para el tratamiento por vía oral en monoterapia de pacientes adultos y pediátricos con tumores sólidos que presentan una fusión del gen NTRK, con una enfermedad localmente avanzada, metastática o cuya resección quirúrgica probablemente genere una elevada morbilidad, y con ausencia de opciones terapéuticas satisfactorias.
Su actividad antitumoral ha sido evidenciada en los modelos tumorales in vitro e in vivo (modelo de ratón) empleados en su desarrollo, incluidas células con activación constitutiva de las proteínas TRK resultantes de fusiones de genes o deleción de un dominio regulador proteico, o en células con sobreexpresión de proteínas TRK. Igual que para entrectinib, se han observado diversas mutaciones de resistencia adquirida a inhibidores de TRK: larotrectinib presentó actividad mínima en líneas celulares con mutaciones puntuales en el dominio cinasa de TRKA (como la mutación G595R), y en el dominio de la cinasa TRKC (como las mutaciones G623R, G696A y F617L). También se desconocen todavía las causas moleculares de la resistencia primaria a este fármaco o si la presencia de un controlador concomitante oncogénico afecta a la eficacia de su inhibición sobre TRK (EMA, 2019; AEMPS, 2023).
Aspectos moleculares
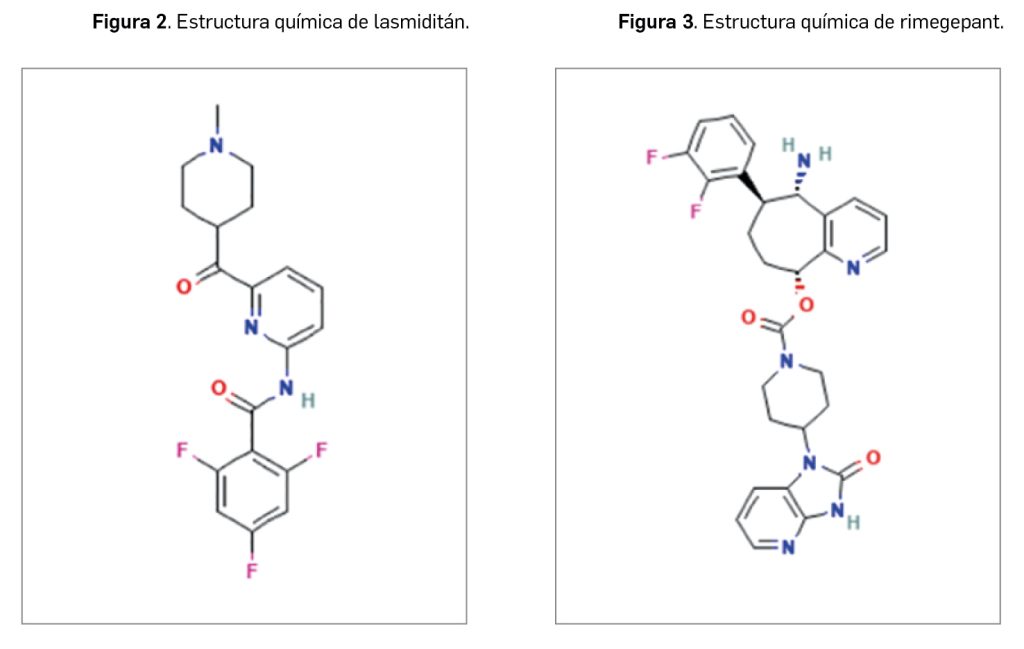
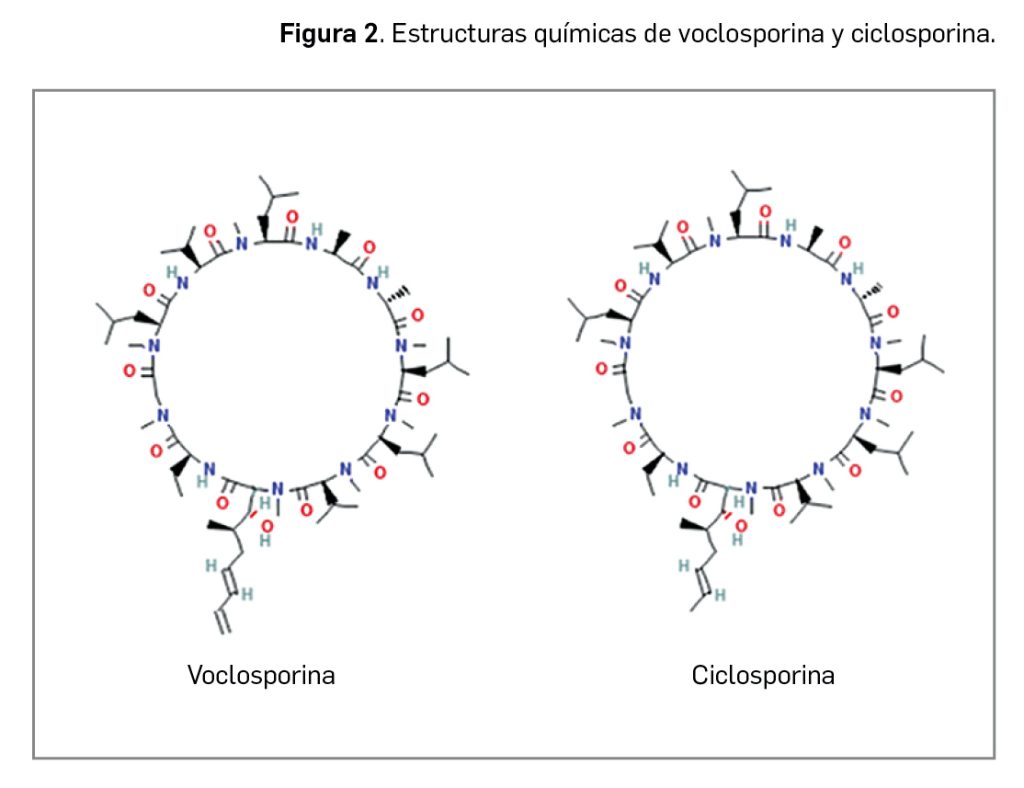
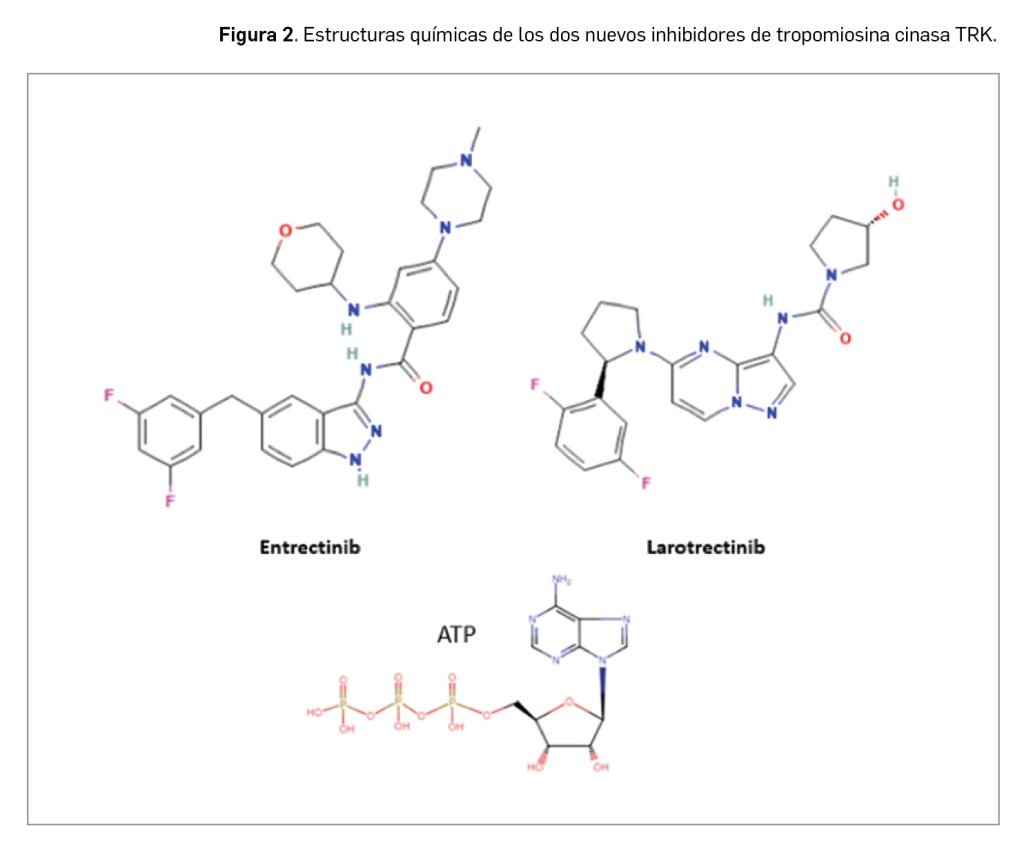
Desde un punto de vista estructural, entrectinib es el N-{5-[(3,5-difluorofenil)metil]-1H-indazol-3-il}-4-(4-methylpiperazin-1-il)-2-[(oxan-4-il)amino]benzamida, que se corresponde con la fórmula C31H34F2N6O2 y un peso molecular relativo de 560,6 g/mol. Con una estructura no quiral (Figura 2), el principio activo se presenta como un polvo no higroscópico de color blanco a blanquecino o rosa pálido, que es pobremente soluble en medio acuoso, pues, al ser una base libre, su solubilidad es altamente dependiente de pH: tiene mayor solubilidad cuanto más bajo es el pH.
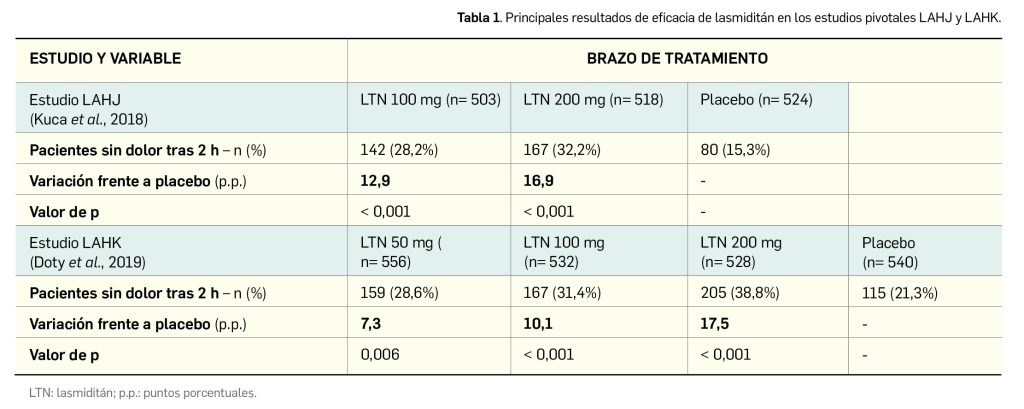
Por su parte, larotrectinib, presente en el medicamento en su forma sulfato, tiene como nombre químico el de (3S)-N-{5-[(2R)-2-(2,5-difluorofenil)-1-pirrolidinil]pirazolo[1,5-a]pirimidin-3-il}-3-hidroxi-1-pirrolidina carboxamida, que se corresponde con la fórmula C21H24F2N6O6S y tiene un peso molecular de 526,5 g/mol. Con una estructura quiral (la molécula tiene estereoisomería por la presencia de dos centros quirales), se presenta como un polvo no higroscópico de color blanquecino a amarillo o amarillo rosado, con una solubilidad baja en la mayoría de los disolventes orgánicos a excepción de los alcoholes (específicamente metanol y etanol) y dependiente de pH en medios acuosos. Los estudios in vitro sugieren que larotrectinib es completamente soluble en todo el rango de pH del tracto gastrointestinal.
Ambos fármacos están estrechamente relacionados estructural y farmacológicamente entre sí y con otros miembros de la serie de inhibidores de proteína cinasas, en la que se encuadran un gran número de principios activos comercializados en España, resultantes de la optimización funcional mediante modelización molecular a partir de una serie de 2-fenilaminopirimidinas, de donde surgió el imatinib, cabeza de serie del grupo. Aunque se aprecia una diversidad estructural importante en el grupo, todos presentan heterociclos nitrogenados y guardan –en mayor o menor grado– una familiaridad química con la molécula de ATP (o, en su caso, con la de GTP, como sucede en las cinasas MAPK), con la cual algunos de los principios activos compiten para provocar el bloqueo de la cinasa correspondiente. Se han desarrollado modelos moleculares de relación estructura-actividad para este grupo de fármacos y, en todos los casos, las interacciones estéricas y electrostáticas han demostrado ser determinantes para el efecto inhibitorio sobre la tirosina cinasa.
Eficacia y seguridad clínicas
ENTRECTINIB
La eficacia y la seguridad de entrectinib en su pauta e indicaciones aprobadas han sido contrastadas en tres ensayos clínicos que han enrolado una población de pacientes adultos con tumores sólidos avanzados o metastásicos y reordenamiento en los genes NTRK, ROS1 o ALK: dos ensayos abiertos fase 1 –de búsqueda de dosis– y sin comparador (estudios ALKA y STARTRK-1) y otro ensayo abierto de fase 2 a dosis fija (estudio STARTRK-2), multicéntrico y no controlado, en el que los pacientes podían haber recibido tratamiento previo y debían presentar un buen estado general8 (ECOG ≤ 2) y enfermedad medible según criterios RECIST v1.1. Además, el fármaco se investigó en población pediátrica en un estudio de fase 1/2 (estudio STARTRK-NG).
El análisis integrado de los 3 estudios de un solo brazo en adultos incluyó inicialmente datos de 93 pacientes (AEMPS, 2022d) y ha sido actualizado en la ficha técnica del medicamento (AEMPS, 2022a) con un corte de datos de hasta 150 sujetos, de los cuales la gran mayoría estaban enrolados en el estudio STARTRK-2. Para incluirse en este análisis no preespecificado, los datos debían proceder de pacientes mayores de 18 años, con un tumor sólido no resecable o metastásico con confirmación de la presencia de genes de fusión NTRK y diagnóstico primario extracraneal, y recibir al menos una dosis diaria de entrectinib de ≥ 600 mg durante al menos 12 meses de seguimiento, no habiendo recibido previamente otro inhibidor de NTRK. En general, el tratamiento se mantuvo hasta la progresión de la enfermedad o aparición de toxicidad inaceptable, siendo la mediana global de la duración del seguimiento de 31 meses.
Entre las características demográficas basales de los pacientes incluidos se deben destacar las siguientes: hubo equilibrio de géneros (49% hombres y 51% mujeres), la mediana de edad fue de 59 años (rango 21-88), la raza predominante era la blanca (59%, 26% de asiáticos y 5% de hispanos), la mayoría de los pacientes tenía buen estado funcional (41% ECOG 0 y 50% ECOG 1) y no tenía antecedentes de tabaquismo (63%). En cuanto a su enfermedad, casi todos tenían metástasis (más de la mitad en pulmón y ganglios linfáticos), el 81% y el 61%, respectivamente, habían recibido con anterioridad cirugía y radioterapia frente a su tumor, y el 77% había recibido terapia sistémica (sobre todo quimioterapia); si bien un tercio de los pacientes no había recibido tratamiento sistémico en el contexto metastásico. Los tumores más frecuentes fueron: sarcoma (21%), cáncer de pulmón (21%), tumores de las glándulas salivales (17%), cáncer de tiroides (11%), cáncer colorrectal (7%), y cáncer de mama (6%).
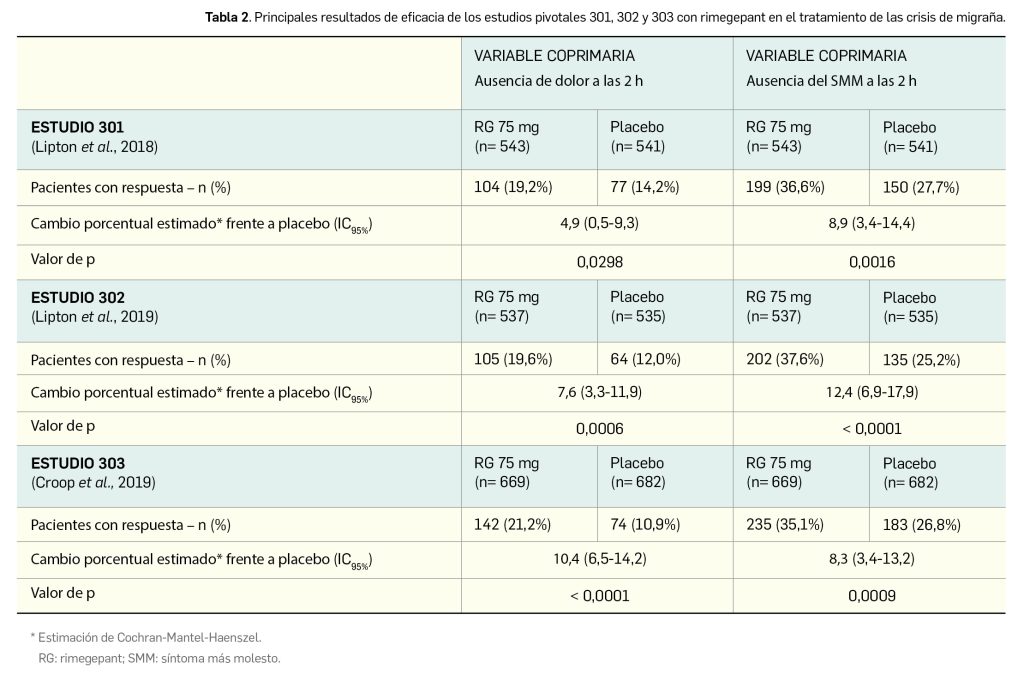
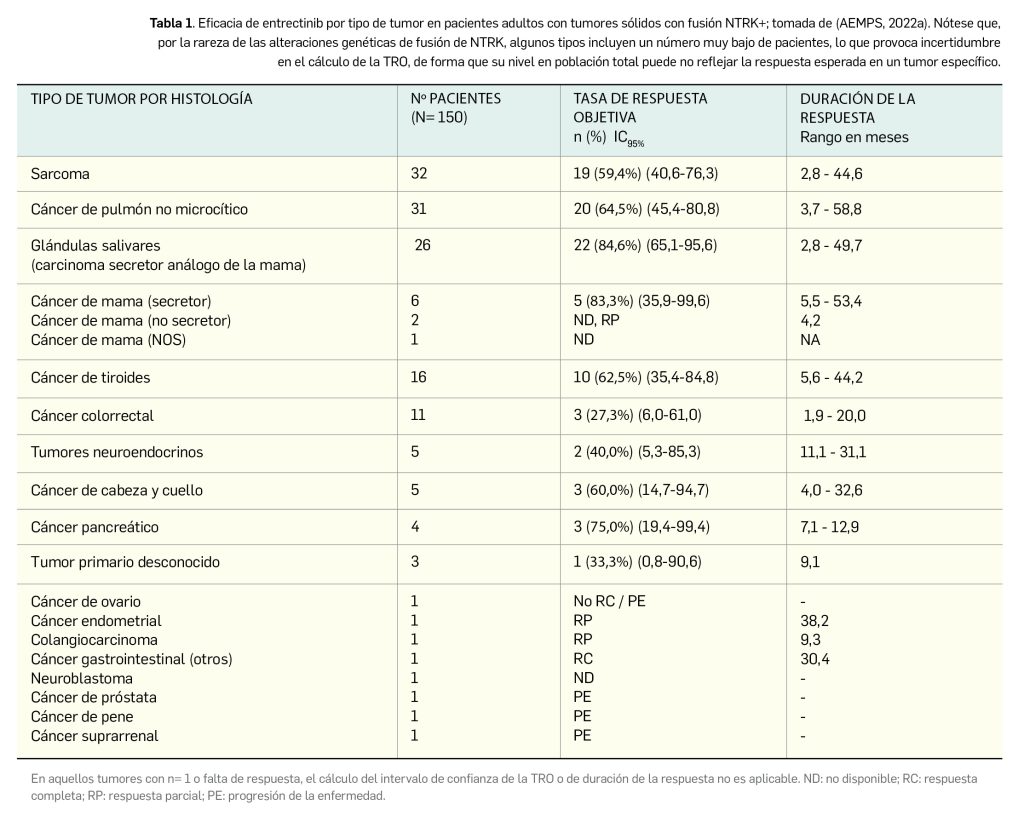
Los resultados tras revisión centralizada y ciega por un comité independiente revelan una tasa de respuesta objetiva (TRO)9–variable primaria– del 61,3% (IC95% 53,0-69,2), con una respuesta verificada en 92 de los 150 pacientes: fue completa en 25 de ellos (16,7%) y parcial en 67 (44,7%). Tras registrarse 50 eventos de progresión, se estableció una mediana de duración de la respuesta –variable secundaria clave– de 20 meses (IC95% 13,2-31,1); así, se observó una respuesta de al menos 6 meses de duración en el 83% de los pacientes, de al menos 9 meses en el 77% y de al menos 12 meses en el 66%. Por tipo histológico de tumor se han publicado los resultados individuales recogidos en la Tabla 1, poniendo de manifiesto que se pueden beneficiar en mayor medida del tratamiento aquellos pacientes que presentan algunos tipos tumorales específicos, como el de glándulas salivares o el cáncer de mama secretor.
En cualquier caso, se vio que en los pacientes con una amplia caracterización molecular antes del tratamiento (n= 78), la TRO general fue del 54%; mayor entre los que no presentaban otras alteraciones genómicas adicionales (n= 17), alcanzando el 77%, respecto a quienes tenían otras mutaciones adicionales distintas a NTRK (n= 61), que mostraron una TRO del 48%. Además, se identificó un subgrupo de 13 pacientes adultos con metástasis medibles en el SNC al inicio (5 habían sido pretratados con radioterapia cerebral en los 2 meses anteriores), en quienes la tasa respuesta intracraneal alcanzó el 69% (3 respuestas completas y 6 parciales) y la mediana de duración de la respuesta, los 17 meses.
La eficacia de entrectinib en pacientes pediátricos de ≥ 12 años se dio por confirmada en base a la extrapolación de los datos de los tres ensayos abiertos con adultos y, sobre todo, por la evidencia derivada del estudio STARTRK-NG. Con un mínimo de 6 meses de seguimiento, la evaluación por el comité ciego independiente en 5 pacientes pediátricos (3 con tumores sólidos y 2 con tumor primario en el SNC) mostró dos respuestas completas (glioblastoma y fibrosarcoma infantil) y 3 respuestas parciales (glioma de alto grado, fibrosarcoma infantil y melanoma metastásico), aunque no se puede concluir porque la mayoría de las respuestas estaban en curso en el momento del corte de datos.
Por otra parte, el nuevo fármaco ganó aprobación en pacientes adultos con CPNM avanzado ROS1+ en base a los resultados de un subanálisis con datos de pacientes incluidos en los tres citados ensayos multicéntricos ALKA, STARTRK-1 y STARTRK-2. En concreto, se consideraron 168 pacientes (35% varones, 44% caucásicos y 45% hispanos) con CPNM recurrente o metastásico y confirmación molecular de mutación en ROS1, quienes debían tener buen estado funcional (41% ECOG 0 y 49% ECOG 1), enfermedad medible según criterios RECIST v1.1, tratamiento diario con el fármaco y seguimiento ≥ 12 meses y no haber recibido terapia previa con un inhibidor de ROS1. Estos pacientes presentaron una mediana de edad de 54 años (rango 20-86), casi dos de cada tres (63%) no tenía antecedentes de tabaquismo, un 37% no había recibido tratamiento sistémico previo y la práctica totalidad (98%) tenía metástasis al inicio (de localización mayoritaria en pulmón, ganglios y cerebro).
Con una duración mediana del seguimiento de 29 meses (actualización de los datos publicados por Drilon et al., 2022), la evaluación ciega por un comité central independiente reportó una tasa de respuesta objetiva del 68% (IC95% 60,2-74,8), solo completas en el 8,7% de los casos, y una mediana de duración de la respuesta de 20,5 meses, cifras que se sitúan muy cercanas a las descritas para la población de tumores sólidos NTRK+. Entre otras variables secundarias, sobresale una mediana de supervivencia libre de progresión de 15,7 meses y de supervivencia global de 47,8 meses, de modo que tras 1 año estaban vivos el 81% de los pacientes. Además, entre los 25 sujetos con metástasis medibles en el SNC al inicio, la tasa de respuesta intracraneal fue del 80% (solo 3 respuestas completas de 19), con una mediana de duración de la respuesta de 13 meses y de supervivencia sin progresión a ese nivel de 8,8 meses.
Desde el punto de vista de la seguridad, se dispone de los datos de los tres estudios en adultos y uno pediátrico, que totalizan más de 500 pacientes expuestos una mediana de 5,5 meses al fármaco (AEMPS, 2022a), y apuntan a que entrectinib tiene un perfil toxicológico importante, similar al de otros inhibidores de cinasas usados en oncología y que determina una importante incidencia de eventos adversos durante el tratamiento (> 90%). No obstante, de las 20 muertes vistas durante el tratamiento, ninguna fue considerada por los investigadores como relacionada con el fármaco.
Las reacciones adversas a entrectinib descritas con mayor frecuencia (> 20%) son las siguientes: disgeusia (41%), cansancio (28%), mareo (25%), estreñimiento (24%), diarrea (23%), náuseas (21%), parestesia, disnea, anemia, aumento de peso, edema, aumento de creatinina en sangre, dolor, trastornos cognitivos (más frecuentes en pacientes con afectación del SNC), vómitos, tos y pirexia. Si bien fueron mayoritariamente leves-moderadas en severidad, entre las reacciones adversas graves destacan la infección pulmonar (5,2%), disfonía (4,6%), trastorno cognitivo (4%), derrame pleural (3,0%) y fracturas (3,8%). Por ello, la tasa de interrupciones temporales (46%) y de abandonos del tratamiento por motivos de seguridad (4%) no es desdeñable. No se han visto grandes diferencias en el perfil de seguridad de entrectinib según grupos etarios, solo destacable la mayor incidencia en niños de neutropenia (28%), aumento de peso (22%), fracturas óseas (11%) y dolor de cabeza (6%). Tampoco se ha evaluado en mujeres embarazadas por el riesgo presupuesto de alteraciones fetales.
LAROTRECTINIB
La eficacia y la seguridad de larotrectinib en su pauta oral diaria e indicación aprobadas se ha evaluado –y se continúa evaluando– en tres ensayos clínicos multicéntricos, abiertos y de un solo grupo que han incluido un total de 313 pacientes adultos y pediátricos con tumores sólidos localmente avanzados o metastásicos de heterogénea histología: un fase 1 y otro fase 1/2 (estudios SCOUT; N= 13 adultos y N= 121 niños y jóvenes de entre 1 mes y 21 años), ambos de búsqueda de dosis, y un tercer ensayo de fase 2 (estudio NAVIGATE; N= 179 adultos y niños mayores de 12 años). Mientras que en los dos primeros se reclutaron pacientes con y sin confirmación de fusión de genes NTRK (en la fase de expansión sí se requirió confirmación), tal confirmación fue criterio de inclusión del estudio NAVIGATE.
De nuevo, se realizó un análisis primario combinado de eficacia en que un comité central de revisión independiente y ciega evaluó10 los datos de 272 pacientes tratados con larotrectinib en los tres estudios, quienes debían presentar fusión positiva de NTRK (la gran mayoría se confirmó por técnicas de secuenciación de nueva generación), enfermedad medible y tumor primario externo al SNC. Para ser incluidos debían haber recibido terapia estándar para su tipo de tumor y estadio, haber sido sometidos a cirugía radical, o tener una alta probabilidad de no tolerancia o beneficio con los cuidados estándar disponibles. Entre sus características basales destaca una mediana de edad de 41 años (rango 0-90), un 57% eran de raza blanca, un 49% hombres y casi todos tenían un buen estado general (ECOG de 0 o 1 en el 89%) y habían recibido tratamiento previo para su tumor (92% cirugía, radioterapia o terapia sistémica); solo un 26% de los pacientes eran naïve al tratamiento sistémico. Los tumores más frecuentes fueron el sarcoma de partes blandas (25%), el fibrosarcoma infantil (18%) y el cáncer de tiroides (11%).
Otros 41 pacientes con tumores primarios del SNC con fusión NTRK+ (astrocitoma, ganglioglioma, glioblastoma, glioma, etc.) y enfermedad basal medible conformaron la población para evaluar la respuesta tumoral en sistema nervioso, todos los cuales –excepto uno– habían recibido tratamiento oncológico previo (cirugía, radioterapia y/o terapia sistémica previa). Entre ellos, la mediana de edad fue de 11 años (rango 1-79), dos tercios eran de raza blanca, la mitad varones y en general presentaban también buen estado funcional (88% puntuación ECOG de 0 o 1).
Habida cuenta de que el tratamiento con larotrectinib se pudo mantener incluso ante progresión de la enfermedad siempre que hubiera beneficio clínico, se han reportado los siguientes resultados para una duración mediana del tratamiento de casi 20 meses y seguimiento aún en curso (Hong et al., 2020; AEMPS, 2023):
-La tasa de respuesta global11–variable principal del estudio NAVIGATE– fue del 67% (182/272; IC95% 61-72) en la población del análisis primario, con un 23% de respuestas completas y un 39% de respuestas parciales; de modo interesante, 13 pacientes tuvieron respuesta patológica completa, es decir, pudieron someterse después del tratamiento con larotrectinib a resección quirúrgica sin células tumorales viables y con márgenes negativos. El nivel de respuesta se redujo ligeramente hasta el 61% (191/313; IC95% 55-66) si se consideraban todos los tumores sólidos NTRK+, incluidos los tumores primarios del SNC, con un 20% de respuestas completas y 37% de respuestas parciales.
- La mediana de la duración de la respuesta –variable co-principal del análisis– fue de 43,3 meses (intervalo 0-65) entre los pacientes con tumores extracraneales, de modo que el 80% de los pacientes tenía una respuesta de duración ≥ 12 meses y el 54% de ≥ 36 meses. Esas cifras fueron similares si se incluían los pacientes con tumores primarios del SNC, si bien la duración global de la respuesta decrecía levemente hasta 41,5 meses.
- La mediana del tiempo transcurrido hasta la primera respuesta fue de 1,8 meses (intervalo 0,9-22,9).
- Sin haberse alcanzado la mediana de supervivencia global en la población de pacientes con tumores externos al SNC, se verificó que al año estaban vivos el 86% y a los 3 años, el 72%; la mediana de supervivencia libre de progresión fue de 30,8 meses, con hasta el 65% de pacientes libres de progresión al año, y el 43% a los 3 años.
- Por tipo de tumor según histología (Tabla 2), la tasa de respuesta a larotrectinib fue variable, de especial relevancia clínica (> 80%) en algunos casos como el fibrosarcoma infantil, el tumor de glándulas salivales o el tumor de mama secretor.
Los análisis posteriores por subgrupos ponen de manifiesto que la eficacia del fármaco puede ser superior en pacientes pediátricos (n= 94), donde larotrectinib logró una tasa de respuesta global del 84%, en comparación con la subpoblación de adultos (n= 178), en quienes la respuesta fue del 58%. De igual modo, los pacientes sin otras alteraciones genómicas distintas a NTRK (n= 110) pueden beneficiarse en mayor medida que los que presentan otras mutaciones adicionales (n= 128): las tasas de respuesta fueron del 76% y el 52%, respectivamente. En todos aquellos que se demostró que no tenían fusión NTRK no se observó respuesta.
La caracterización de la seguridad de larotrectinib se basa en los datos de 335 pacientes con tumores NTRK+ en los tres ensayos clínicos principales y en la evidencia poscomercialización, con tratamientos de una duración mediana de 15 meses. El perfil toxicológico del fármaco parece importante pero clínicamente manejable: casi todos los pacientes notifican algún evento adverso, que se relaciona con el fármaco en casi 4 de cada 5 casos. Pero la mayoría son leves-moderadas en severidad y ninguna de las muertes registradas en los estudios se asoció al tratamiento.
Las reacciones adversas a larotrectinib más frecuentemente reportadas (> 20%) fueron aumento de transaminasas (31-33%), vómitos (28%), anemia (27%), estreñimiento (27%), diarrea (25%), náuseas (23%), fatiga (22%) y mareo (20%). Los casos considerados graves fueron muy escasos (≤ 2%), describiéndose algunas como neutropenia y leucopenia, aumento de transaminasas, debilidad muscular o reacciones neurológicas (mareo, ataxia, parestesia). Esa baja incidencia de reacciones adversas graves determinó una baja tasa de suspensión permanente del tratamiento por motivos de seguridad, que fue del 2%; hubo mayor frecuencia de reducciones de dosis por tal causa, pero la mayoría de los casos fueron en los primeros tres meses de tratamiento. Como con entrectinib, tampoco con larotrectinib se observaron diferencias sustanciales en el perfil de seguridad por grupos etarios, solo destacable la mayor incidencia de reacciones adversas graves en pacientes de menos de 6 años de edad, que se notificaron en hasta el 69% de los niños de hasta 3 meses y en el 48% desde los 3 meses, destacando la neutropenia.
Aspectos innovadores
Entrectinib y larotrectinib son dos nuevos fármacos de molécula pequeña activos por vía oral que ejercen una actividad inhibitoria de tirosina cinasas, competitiva con ATP y selectiva para los receptores de la tropomiosina cinasa TRK (TRKA, TRKB y TRKC); entrectinib además inhibe la tirosina cinasa ROS1 y la cinasa del linfoma anaplásico (ALK). Todas ellas son proteínas con potencial tumorigénico que, cuando se encuentran sobreactivadas por mutación en sus genes, determinan la hiperactivación de las vías de señalización descendentes (como las de MAPK o PI3K/AKT) que da lugar a una proliferación celular ilimitada.
Se comprende así que la inhibición que ejercen los nuevos fármacos sobre ellas se traduzca en un bloqueo o reducción de la proliferación celular y la inducción de la apoptosis de las células tumorales, en base a lo cual sus medicamentos han recibido autorización condicional para el tratamiento por vía oral en monoterapia de pacientes adultos y pediátricos con tumores sólidos que presentan una fusión del gen NTRK y tienen enfermedad localmente avanzada, metastática o cuya resección quirúrgica probablemente genere una elevada morbilidad, y con ausencia de opciones terapéuticas satisfactorias. La autorización de entrectinib se ha acotado a mayores de 12 años y siempre que no hayan recibido previamente un inhibidor de TRK, pero ha ganado indicación adicional como tratamiento en monoterapia de pacientes adultos con cáncer de pulmón no microcítico avanzado ROS1+ no tratados previamente con inhibidores de ROS.
Por orden cronológico, larotrectinib fue el primer inhibidor del receptor de la tropomiosina cinasa (TRK) diseñado racionalmente para evitar la actividad sobre otras enzimas, y el primero en aprobarse en la UE y en comercializarse en España con una indicación agnóstica, o sea, sin especificar el tipo o localización del tumor, sino que valdrá para tratar cualquier tumor con fusión de genes NTRK. Su autorización en pauta oral diaria se sustentó en los resultados de eficacia de un análisis combinado de 3 ensayos aún en marcha en fases tempranas de la investigación clínica y heterogéneos en diseño (abiertos, multicéntricos y de un solo brazo), destacando por su tamaño el estudio fase 2 NAVIGATE; en conjunto enrolaron a 313 pacientes adultos y pediátricos con tumores sólidos localmente avanzados o metastásicos de diversa histología y localización.
Con una duración mediana del tratamiento con larotrectinib de casi 20 meses, en pacientes con afectación extracerebral primaria se alcanzó una tasa de respuesta global del 67% (23% de respuestas completas, 7% con respuesta patológica completa) y en quienes tenían tumor primario en el SNC, esa tasa fue del 61% (20% de respuestas completas). De modo interesante, su efecto antitumoral es rápido (mediana de tiempo hasta la respuesta de < 2 meses) y duradero, con una mediana de duración de la respuesta de 43 meses, similar si se consideran también los tumores intracraneales. Se verificó que al año de tratamiento estaban vivos el 86% de los pacientes con tumores primarios fuera del SNC y, a los 3 años, el 72%, no habiéndose alcanzado la mediana de supervivencia global, la variable más robusta en oncología; la mediana de supervivencia sin progresión fue de casi 31 meses.
Los datos apuntan a una mayor eficacia del fármaco en pacientes pediátricos (tasa respuesta del 84% vs. 58% en adultos) y en quienes no presentan otras mutaciones adicionales (76% vs. 52% de pacientes con otras alteraciones genéticas). Por tipo de tumor según histología, la respuesta al nuevo fármaco fue variable, de especial relevancia en algunos tumores raros con alta frecuencia de fusión de NTRK, como el nefroma mesoblástico congénito (100%), el fibrosarcoma infantil (92%), el tumor de glándulas salivales (84%), el tumor de mama secretor o el de estroma gastrointestinal (80%).
Larotrectinib tiene un perfil de seguridad importante, en línea con otros inhibidores de cinasas, pero clínicamente manejable: pese a una alta notificación de eventos adversos relacionados con su uso (80%), casi todos son leves-moderados en severidad, y ninguno fatal. Las reacciones adversas más frecuentes (≥ 20%) fueron: fatiga, hepatotoxicidad con aumento de transaminasas, mareo, alteraciones digestivas (estreñimiento, náuseas y vómitos) o anemia; las reacciones adversas graves fueron poco comunes (≤ 2%), destacando las citopenias, aumento de transaminasas, debilidad muscular o reacciones neurológicas (mareo, ataxia, parestesia). Así, aunque la modificación de pauta posológica fue frecuente, la tasa de suspensión definitiva del tratamiento por motivos de seguridad fue menor del 4%. En niños de < 6 años, habrá que tener especial precaución con la neutropenia. No obstante, se requiere conocer en mayor medida la seguridad del fármaco a largo plazo (> 2 años).
Se pueden identificar otras limitaciones notables en la evidencia disponible, entre las que la mayor incertidumbre se produce por la carencia de un grupo control, el uso de variables subrogadas (tasa de respuesta) y la inmadurez de los datos por la continuación de los estudios; la escasa muestra de pacientes para algunos tipos tumorales también complica las conclusiones particulares para cada tipo de cáncer a partir de la tasa de respuesta global, si bien la EMA consideró aceptables las poblaciones incluidas por la dificultad de reclutamiento dada la baja prevalencia de tumores con genes de fusión NTRK (0,3-1% globalmente). Además, aunque el EPAR (EMA, 2019) recoge los resultados disponibles para las terapias sistémicas más frecuentemente usadas en práctica cínica real de los tumores estudiados, la ausencia de estudios comparativos directos con larotrectinib y la variabilidad de las poblaciones –no superponibles– evaluadas (complica cualquier comparación indirecta entre ellas) impiden concluir sobre el beneficio incremental (y su magnitud) que aporta en términos de supervivencia respecto a otras opciones de tratamiento. En esa línea, es preciso citar que el IPT incluye una orientación sobre la relevancia del beneficio clínico que puede aportar según una escala de la Sociedad Europea de Oncología Médica, que sugiere un beneficio moderado (puntuación de 3 en una escala de 1 a 5), aunque en estudios de un solo brazo esta valoración es poco fiable.
Poco después de larotrectinib (un mes después, también en 2023) se comercializó en España entrectinib, el segundo inhibidor de TRK disponible en España, diseñado específicamente para atravesar la barrera hematoencefálica (es sustrato débil de la glicoproteína P) a fin de abordar también la localización intracraneal de los tumores con fusiones de genes NTRK. Su desarrollo clínico fue similar al de larotrectinib y su aprobación basada en el análisis integrado de 3 ensayos clínicos de fase 1 y 2, no controlados y de un solo brazo; se dispone de datos de hasta 150 pacientes adultos con tumor avanzado con fusión NTRK+ y localización primaria extracraneal, la mayoría de los cuales se incluyeron en el estudio STARTRK-2, un estudio multicéntrico de fase 2.
Sus resultados revelan que el tratamiento diario con entrectinib aporta una tasa de respuesta global del 61% (completa en el 17% de los casos), y una mediana de duración de la respuesta de 20 meses, de forma que permanecen con respuesta tras al menos 1 año dos tercios de los pacientes respondedores. Con una tasa de respuesta (69%) y duración (17 meses) parecidas en los pacientes con metástasis intracraneales, todo apunta de nuevo a una mayor eficacia entre los que no presentaban otras alteraciones genómicas adicionales (77% vs. 48% con mutaciones adicionales distintas a NTRK). Por tipo histológico, los datos también sugieren que el beneficio con entrectinib es mayor en los pacientes que presentan algunos tipos tumorales específicos, como el de glándulas salivares, el cáncer de mama secretor o el pancreático, en que alcanzan respuestas superiores de ≥ 75%. No obstante, las limitaciones de la evidencia son similares a las descritas para larotrectinib e impiden también sacar conclusiones definitivas sobre el beneficio incremental que aporta sobre otras opciones terapéuticas.
Adicionalmente, la autorización del fármaco en CPNM avanzado ROS1+ derivó de un subanálisis de los datos de 168 pacientes con tumor recurrente o metastásico no pretratados con inhibidores de ROS, que mostró una tasa de respuesta objetiva del 68% (solo 9% completas) y una mediana de duración de la respuesta de 21 meses, las cuales resultan en una mediana de supervivencia sin progresión de 16 meses y de supervivencia global de 48 meses, de modo que tras 1 año siguen vivos > 80% de los pacientes. Entre los que tenían metástasis cerebrales, el fármaco aportó una respuesta intracraneal del 80%, con una mediana de SLP a ese nivel de 9 meses.
En ambas indicaciones, el perfil de seguridad de entrectinib es consistente y similar al comentado para larotrectinib, sobresaliendo por su frecuencia (> 10%) las siguientes reacciones adversas: alteraciones del tracto gastrointestinal (disgeusia, estreñimiento, diarrea, náuseas y vómitos), fatiga y dolores musculo-articulares, y alteraciones del perfil bioquímico sugerentes de hepatotoxicidad (elevación de creatinina y transaminasas). En su mayoría son eventos adversos leves-moderados, si bien entre las reacciones graves –pasajeras tras la suspensión del tratamiento– destacan infección pulmonar (5%), disfonía (5%), trastorno cognitivo (4%), derrame pleural (3%) y fracturas óseas (4%). La neutropenia y las fracturas óseas parecen más frecuentes en niños.
Asumiendo la ausencia de comparaciones con las alternativas empleadas actualmente en cada tipo de tumor, no se pueden establecer diferencias entre entrectinib y larotrectinib en el tratamiento de aquellos cánceres con fusiones NTRK+, aunque a la vista de los datos no parece que entrectinib comporte ninguna ventaja sobre su predecesor (por ejemplo, larotrectinib aporta mayor tasa de respuesta y duplica su duración, y cubre a toda la población pediátrica), debiéndose diferenciar el beneficio que puedan aportar ambos en aquellas poblaciones con escasas alternativas terapéuticas de aquellos tumores que disponen de tratamientos con eficacia demostrada. Tampoco se puede establecer la superioridad de entrectinib frente a crizotinib en pacientes con CPNM ROS1+ (no se han realizado ensayos comparativos ni son posibles por ahora comparaciones indirectas ajustadas12), considerándose ambos como opciones de tratamiento adecuadas y de uso preferente en primera línea frente a la quimioterapia (AEMPS, 2022d)13.
Aunque parece haber consenso en torno a la necesidad de disponer de una evidencia más sólida para recomendar de forma generalizada el uso de estos nuevos fármacos, algunas sociedades científicas (Yoshino et al., 2020), autoridades sanitarias y agencias reguladoras a nivel internacional, como el NICE británico o las agencias canadiense o italiana, ya recomiendan –con restricciones– los inhibidores de TRK como opción preferible para el tratamiento dirigido de pacientes con tumores sólidos portadores de una fusión positiva de NTRK. De forma previa resultará esencial hacer hincapié en las pruebas de diagnóstico que evidencien la alteración genética en el tumor y la selección de pacientes.
En resumen, las poblaciones candidatas al tratamiento con laro- y entrectinib en la práctica son más amplias que las incluidas en los ensayos clínicos (para algunos tipos tumorales no hay ningún dato de eficacia), por lo que la valoración de ambos fármacos en el marco pantumoral es compleja debido a los múltiples factores pronósticos intrínsecos a cada una de las histologías tumorales investigadas además de la variedad de abordajes terapéuticos. En cualquier caso, por la falta de alternativas eficaces en la gran mayoría de los tumores NTRK+ en estadios avanzados la magnitud de los resultados alcanzados con ambos fármacos se considera positiva y de relevancia clínica.
Estamos ante dos medicamentos que han supuesto un cambio de paradigma en el abordaje de determinados tipos tumorales –del enfoque centrado en histología/localización al centrado exclusivamente en el perfil molecular–, pero que requieren de un procedimiento de estudio genético previo que no se realiza de forma rutinaria en la práctica habitual en España, lo cual hace por ahora desconocido el volumen real de población que se puede beneficiar de su uso y puede mermar su potencial terapéutico, a expensas de que futuros estudios permitan definir mejor su positivo balance beneficio-riesgo, incluido un mayor conocimiento sobre la relevancia de las mutaciones de resistencia a los fármacos y sus causas. Sea como fuere, el número de pacientes que podrán usar larotrectinib y entrectinib será pequeño, por la baja frecuencia (< 1%) de las fusiones NTRK+ en el conjunto de tumores sólidos.