Resumen
Las zoonosis, definidas como patologías infecciosas de etiología diversa transmitidas directa o indirectamente desde animales vertebrados a los seres humanos por distintas vías (contacto directo, transmisión alimentaria, ambiental o a través de vectores), constituyen un importante problema global debido a la estrecha relación del ser humano con los animales en el medio agrícola, la vida cotidiana (animales de compañía) y el entorno natural. No solo afectan a la salud del ser humano, también pueden causar alteraciones en la producción y en el comercio de productos de origen animal destinados a la alimentación y otros usos. La magnitud del problema se refleja en las estimaciones más recientes: las zoonosis afectan globalmente a más de 2.700 millones de personas y provocan la muerte de 2,7 millones de ellas al año, sobre todo en las zonas más desfavorecidas del planeta. Hasta el 75% de las enfermedades emergentes, entre las que se incluiría la COVID-19, tienen origen animal.
La sanidad animal constituye, pues, un factor clave para el desarrollo de la ganadería y es de vital relevancia tanto para la economía mundial como para la salud pública, así como para el mantenimiento y conservación de la diversidad de especies animales. En ese contexto, los medicamentos veterinarios –entre los que destacan las vacunas preventivas– se convierten en una herramienta fundamental para asegurar una buena salud de los animales que redunde en un menor riesgo de la difusión de zoonosis y que, en el marco del enfoque One Health, contribuya a limitar o prevenir posibles futuras pandemias y otros riesgos de salud.
Estos medicamentos deben probar en su desarrollo clínico, igual que los medicamentos de uso humanos, altos estándares de calidad, seguridad y eficacia, si bien tienen ciertas particularidades que vienen definidas, entre otros factores, por: las numerosas especies de destino, estableciéndose diferencias entre los animales productores de alimentos y aquellos que no lo son; ciertas formas farmacéuticas y vías de administración específicas; sus condiciones y lugares de dispensación; o la necesidad de hacer un seguimiento especial de los niveles de consumo de algunos de ellos. El presente artículo revisa estos y otros aspectos relativos a los medicamentos de uso animal y centra el foco, finalmente, en el papel asistencial que el farmacéutico, como profesional sanitario experto en el medicamento, desarrolla en torno a los mismos, con gran potencial en educación sanitaria y en la optimización de los resultados de la farmacoterapia en animales.
La sanidad animal en el enfoque One Health
La Organización Mundial de la Salud (OMS) incluye bajo el término de zoonosis (del griego zoon, “animal”, y nosos, “enfermedad”) o enfermedad zoonótica a todas aquellas patologías infecciosas que se transmiten de forma natural entre los animales vertebrados y los seres humanos. Los patógenos zoonóticos pueden ser bacterias, virus, parásitos o agentes no convencionales (priones) y propagarse a los humanos por contacto directo a partir de un reservorio animal (por ejemplo, la rabia, cuando se entra en contacto con la saliva de un perro infectado) o a través de vías indirectas, como los alimentos o el agua (entre otras, la salmonelosis, la listeriosis o la toxoplasmosis), o a través del medio ambiente (por ejemplo, a través de aerosoles o secreciones respiratorias, en el caso de la histoplasmosis, el ébola, la tuberculosis o la COVID-19). También son zoonosis algunas enfermedades transmitidas indirectamente por vectores –tales como mosquitos o garrapatas, entre otros–, como ha ocurrido en algunas enfermedades de las que se han notificado brotes en España en los últimos años: la enfermedad de Lyme, la fiebre hemorrágica de Crimea-Congo, la fiebre del Nilo Occidental o la leishmaniosis.
Las zoonosis constituyen en su conjunto un importante problema de salud pública en todo el mundo debido a la estrecha relación de los seres humanos con los animales en el medio agrícola, la vida cotidiana (animales de compañía) y el entorno natural, pudiendo llegar a causar alteraciones en la producción y el comercio de productos de origen animal destinados a la alimentación y otros usos.
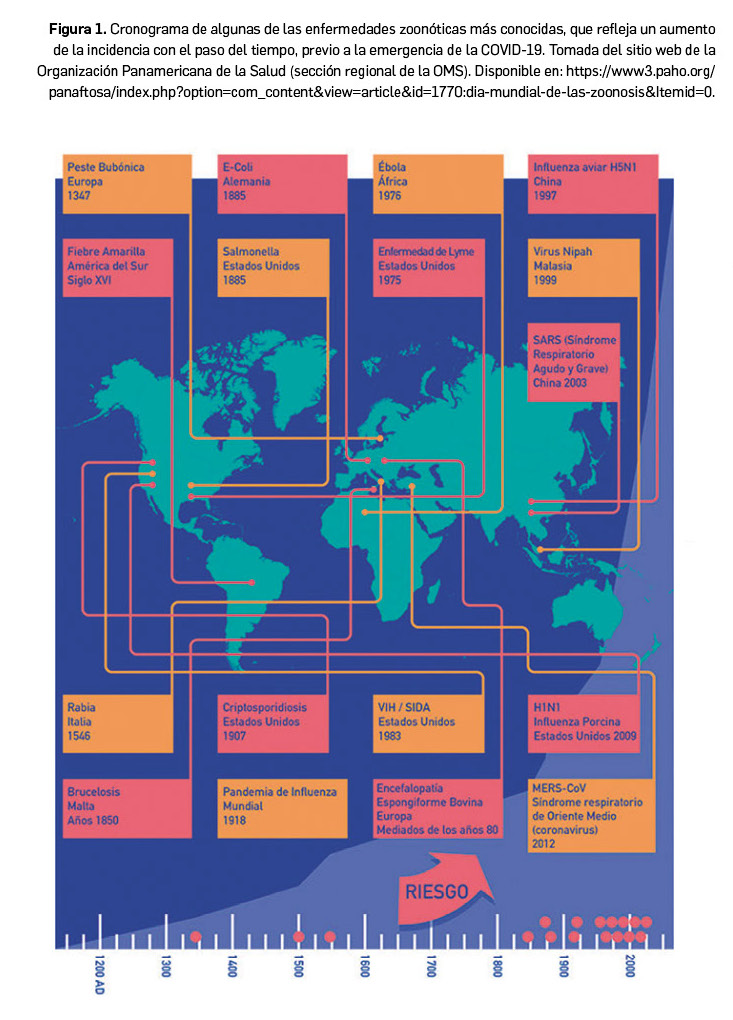
Representan un gran porcentaje de todas las enfermedades infecciosas recientemente identificadas, así como de muchas de las ya existentes. Algunas enfermedades, como la provocada por el VIH, comenzaron como una zoonosis, pero más tarde mutan en cepas exclusivas de los humanos. Otras zoonosis pueden causar brotes recurrentes de enfermedades, como la enfermedad por el virus del Ébola y la salmonelosis. Y otras, como la COVID-19 causada por el nuevo coronavirus SARS-CoV-2, tienen el potencial de causar pandemias mundiales. Su incidencia en clara tendencia creciente a lo largo de la historia (Figura 1) se debe, al menos en parte, al crecimiento de la población, de la globalización, los viajes, el comercio internacional, la mayor tenencia de mascotas, el cambio climático y otros factores socioeconómicos.
La Organización Mundial de Sanidad Animal, también conocida por su nombre original como OIE (Oficina Internacional de Epizootias), ha aportado recientemente las siguientes estimaciones1:
_Un 60% de las enfermedades que afectan al hombre son zoonóticas (de los 1.415 patógenos humanos conocidos en el mundo, hasta 863 –casi 2 de cada 3– se comparten con los animales, es decir, son zoonóticos) y un 75% de los agentes patógenos que originan enfermedades infecciosas emergentes2 del ser humano son de origen animal.
_Cada año, se describen, por término medio, 5 nuevas enfermedades que afectan al hombre, de las cuales 3 son de origen animal.
_Un 85% de los agentes patógenos que se utilizan con fines de bioterrorismo son zoonóticos.
_Incluyendo también las enfermedades diarreicas de transmisión alimentaria o hídrica, las zoonosis afectan en todo el mundo a más de 2.700 millones de personas y provocan de 2,7 millones de muertes al año, sobre todo en las zonas más desfavorecidas del planeta.
Según se ha sugerido, la sanidad animal resulta fundamental para garantizar la salud pública y la seguridad y abastecimiento de los alimentos; es, de igual modo, fundamental para la economía y para el mantenimiento y conservación de la diversidad de especies animales.
No cabe duda de que la base de una buena salud animal se encuentra en la existencia de una adecuada ordenación sanitaria del sector productivo, razón por la que las normas legales vigentes establecen condiciones sanitarias básicas en las explotaciones, el apoyo a la creación de agrupaciones de defensa sanitaria ganadera y la regulación de la calificación sanitaria. Los animales sanos son imprescindibles para la obtención de alimentos seguros, de calidad y a precios razonables que satisfagan las necesidades de la población.
Si bien no todas las enfermedades animales representan un riesgo directo para el ser humano, algunas pueden tener importantes repercusiones socio-económicas, entendiendo que, para muchas personas en el planeta, la sanidad animal no se limita a una cuestión de salud: sus empleos y medios de sustento dependen de ella. Los datos son clarificadores: 1 de cada 5 personas depende directamente de los animales de producción para sus ingresos y medios de sustento; en 2050 será necesario un 70% más de proteína animal adicional para alimentar a toda la población mundial; y más del 20% de las pérdidas de producción animal globales se vinculan con las enfermedades animales.
En este sentido, los gobiernos deben asumir este problema de salud pública con políticas que tomen en cuenta los factores que aumentan el riesgo y dificultan el control de las zoonosis, tales como el cambio climático, la deforestación y los incendios forestales (afectan a la biodiversidad genética de la vegetación y la destrucción del hábitat animal), el incremento de la relación hombre-animales silvestres, los viajes intercontinentales o las mutaciones de los agentes etiológicos con otros genotipos y nuevos vectores transmisores. Los programas sanitarios de prevención coordinados entre las diferentes administraciones y los profesionales sanitarios, así como la disponibilidad de herramientas de control, suponen un elemento clave a la hora de garantizar un elevado nivel de salud pública y de seguridad alimentaria, para reducir al mínimo la incidencia de enfermedades con repercusión en la salud de los consumidores.
Con este fin, la industria farmacéutica ha puesto a disposición de la ganadería potentes y eficaces productos para preservar la sanidad animal: los medicamentos veterinarios. No obstante, estos pueden presentar notorios efectos nocivos para los consumidores de carnes o productos ganaderos cuando son manejados de forma inadecuada, o cuando no se respetan los pertinentes tiempos de espera para que el organismo animal los elimine, razón por la que se impone el control de su aplicación, así como del tiempo de espera de eliminación y el control de los niveles de fármacos en productos destinados al consumo. El uso responsable de los medicamentos asegura los objetivos económicos garantizando el bienestar de los animales productores de alimentos (evita el sufrimiento que pueden padecer por enfermedades en las distintas etapas de su vida) y la salubridad de las carnes y los productos ganaderos en el momento del consumo.
En definitiva, la salud humana y la sanidad animal son estrechamente interdependientes y están vinculadas a todos los ecosistemas en los cuales coexisten. Esto requiere el planteamiento colaborativo global para afrontar la salud como un bien común: el control de los patógenos zoonóticos en su origen animal es la solución más eficaz y económica para proteger al hombre. Esto se ilustra claramente por el hecho de que los antibióticos han representado el mayor avance biomédico en la historia de la humanidad.
Por tanto, como parte del enfoque One Health o Una Única Salud, la OMS se coordina con la Organización de las Naciones Unidas para la Alimentación y la Agricultura (FAO) y la Organización Mundial de Sanidad Animal (OIE) en el Sistema mundial de alerta anticipada ante las principales enfermedades de los animales (GLEWS, por sus siglas en inglés). La crisis sociosanitaria sin precedentes que ha supuesto la pandemia por COVID-19, aún vigente a mediados de 2022, ha puesto de manifiesto la necesidad de impulsar y perseverar en la aplicación de dicho enfoque, que viene a destacar la interrelación entre la salud humana, la salud animal y la salud medioambiental. Con este punto de inflexión a la hora de entender la salud pública, se vuelve prioritario gestionar y controlar las enfermedades de animales, sobre todo aquellas zoonóticas con riesgo de “dar el salto” a los seres humanos. Resulta imprescindible para ello compartir datos epidemiológicos y de laboratorio con el fin de prevenir y detectar brotes de zoonosis y problemas relacionados con la seguridad alimentaria.
Las citadas instituciones colaboran estrechamente para promover respuestas multisectoriales a los peligros para la salud que se manifiestan en relación con: el riesgo de zoonosis, la inocuidad de los alimentos o la orientación sobre reducción de riesgos. Este sistema conjunto se basa en el valor añadido de combinar y coordinar los mecanismos de alerta de los tres organismos para ayudar en la alerta temprana, la prevención y el control de las amenazas de enfermedades animales, incluidas las zoonosis, mediante el intercambio de datos y la evaluación de riesgos, que debe traducirse en medidas regulatorias por parte de las administraciones sanitarias.
Uno de esos principales riesgos, como se verá más adelante, es la resistencia a los agentes antimicrobianos, un factor que complica el control y la prevención de las zoonosis: dado que el uso de antibióticos en los animales criados para la alimentación está muy extendido, aumenta la posibilidad de que aparezcan cepas de patógenos zoonóticos farmacorresistentes capaces de propagarse rápidamente en las poblaciones animales y humana (Zorn et al., 2022).
Los siguientes apartados se centran en el estado actual de distintos aspectos relativos a los medicamentos veterinarios de interés para los profesionales sanitarios.
Medicamentos veterinarios: definición y entorno regulatorio
El panorama del mercado de medicamentos veterinarios viene regulado por normas legislativas distintas a las que rigen en los medicamentos de uso humano, fundamentalmente en lo tocante a su registro y proceso de autorización de comercialización (requerimiento de receta veterinaria y establecimiento de tiempos de espera), que pretende proteger la salud del consumidor de alimentos de origen animal. La Normativa Europea regula qué medicamentos y cómo se pueden administrar para tratar las enfermedades que afectan a los animales productores de alimentos, asegurando que no queden residuos que afecten a la salud del consumidor.
Se debe recordar que los medicamentos veterinarios se definen según el Real Decreto Legislativo 1/2015 (RDL 1/2015), de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios, como “toda sustancia o combinación de sustancias que se presente como poseedora de propiedades curativas o preventivas con respecto a las enfermedades animales o que pueda administrarse al animal con el fin de restablecer, corregir o modificar sus funciones fisiológicas ejerciendo una acción farmacológica, inmunológica o metabólica, o de establecer un diagnóstico veterinario”. También se considerarán medicamentos veterinarios las “premezclas para piensos medicamentosos elaboradas para ser incorporadas a un pienso”.
En el artículo 6 del anterior Real Decreto 109/1995, de 27 de enero, sobre medicamentos veterinarios, se establecen los diferentes tipos de medicamentos veterinarios legalmente reconocidos: i) medicamentos de uso veterinario; ii) medicamentos prefabricados de uso veterinario; iii) premezclas medicamentosas y los productos intermedios elaborados con las mismas con destino a piensos; iv) fórmulas magistrales destinadas a los animales; v) preparados oficinales destinados a los animales; y vi) autovacunas de uso veterinario. Además, dicho Real Decreto contempla la existencia de medicamentos especiales, entre los cuales se incluirían los estupefacientes y psicótropos, los medicamentos con plantas medicinales con destino a los animales, radiofármacos o medicamentos homeopáticos, entre otros.
Sea como fuere, el medicamento veterinario, como medicamento que es, está sujeto a unas estrictas medidas de control técnico y jurídico a lo largo de toda su vida, desde la investigación del mismo a los procesos de fabricación, comercialización, distribución y dispensación. A diferencia de lo que sucede con los medicamentos de uso humano, en los que la dispensación está limitada a la farmacia comunitaria, el RDL 1/2015 contempla en su artículo 38 la posibilidad de la dispensación de medicamentos veterinarios por parte de otros establecimientos, si bien, y salvo determinadas excepciones, esta dispensación debe ser siempre llevada a cabo obligatoriamente con la intermediación de un farmacéutico.
La UE decidió, hace una década, hacer una revisión en profundidad de la legislación comunitaria sobre medicamento veterinarios y tras varios años de discusiones se materializó en la aprobación de dos Reglamentos fundamentales que han entrado en vigor en 2022.
El primero de ellos es el Reglamento (UE) 2019/6 del Parlamento Europeo y del Consejo, de 11 de diciembre de 2018 (publicado en el DOUE el 7 de enero de 2019), sobre medicamentos veterinarios y por el que se deroga la Directiva 2001/82/CE. Su aplicación práctica entró en vigor el 28 de enero de 2022 y ha supuesto un nuevo paradigma en la regulación europea del medicamento veterinario. En España, este Reglamento se ha acompañado de la publicación del Real Decreto 1157/2021, de 28 de diciembre, por el que se regulan los medicamentos veterinarios fabricados industrialmente, y en el cual se establecen, entre otras cuestiones, los requisitos de la solicitud para la autorización de comercialización, los diferentes procedimientos de autorización, la inscripción de los medicamentos en el Registro de medicamentos veterinarios, la farmacovigilancia, el comercio paralelo o la publicidad. De igual modo, en la actualidad se encuentra pendiente la publicación de un Real Decreto que derogue el RD 109/1995, de 27 de enero, sobre medicamentos veterinarios, y que defina aspectos concretos relacionados con la distribución, prescripción y dispensación de estos medicamentos.
En líneas generales, el Reglamento 2019/6 reconoce 3 grandes grupos de medicamentos de uso animal: a) medicamentos veterinarios diferentes a los medicamentos biológicos; b) medicamentos veterinarios biológicos, incluyendo inmunológicos y otros biológicos no inmunológicos; y c) nuevas terapias. Su Anexo II especifica los requisitos que se tienen que cumplir los medicamentos de cada tipo, a fin de que los laboratorios solicitantes puedan completar el dossier que debe presentarse para su registro.
Para registrar un medicamento, ya sea de uso humano o animal, debe demostrarse que este alcanza los estándares de calidad establecidos, que en las condiciones de uso presenta una seguridad suficiente para asegurar que el beneficio supera al riesgo y que tiene la eficacia en las condiciones de uso que indica la información del producto (ficha técnica), que es parte de la autorización y, por tanto, de obligado cumplimiento. En la evaluación de los datos aportados, las agencias reguladoras –la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) a nivel nacional y la Agencia Europea de Medicamentos (EMA) en el ámbito de la UE– valorarán el beneficio (eficacia) y los riesgos, y únicamente se considerará positiva si el beneficio supera al riesgo. Cabe destacar que la evaluación de los riesgos será distinta para medicamentos de uso humano o veterinarios.
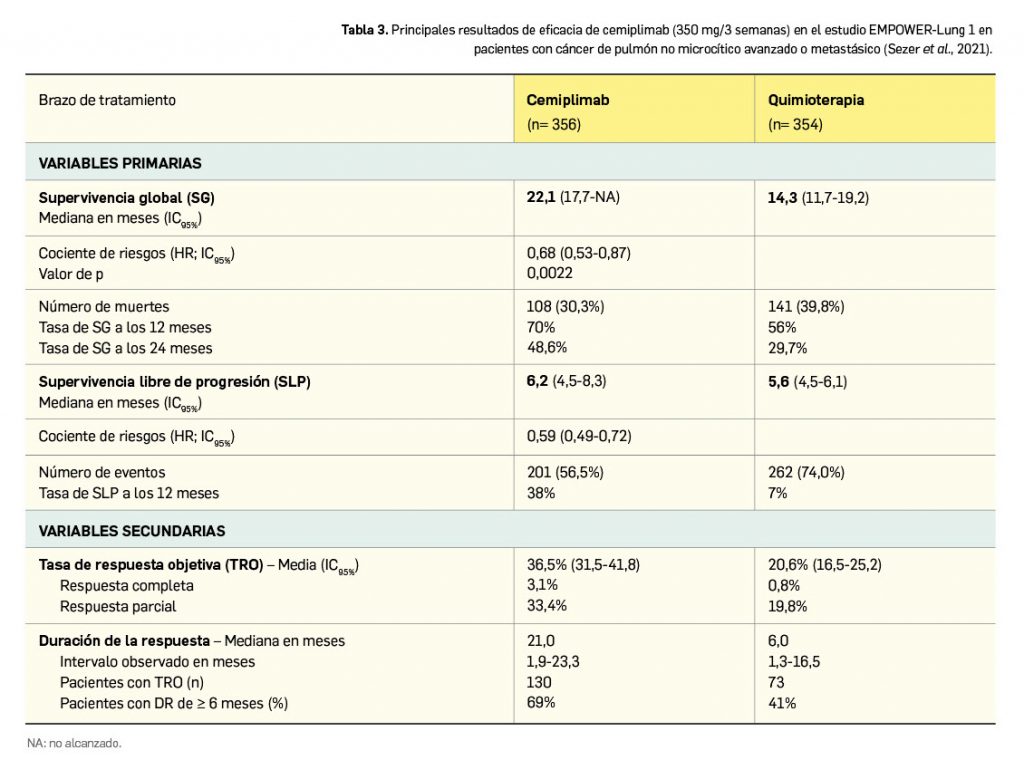
A diferencia de lo que sucede con los medicamentos de uso humano (Tabla 1), para los que existe un acuerdo internacional más amplio, la estructura del expediente de un medicamento veterinario se ha adoptado en la UE y se divide igualmente en 5 partes:
- Sumario del expediente.
- Fisicoquímica, biológica o microbiológica (calidad).
- Seguridad y establecimiento de tiempo de espera (seguridad).
- Estudios preclínicos y clínicos (eficacia).
- Evaluación beneficio/riesgo del medicamento.

Si se comparan ambos modelos, la Parte 2 de veterinaria coincide con el módulo 3 de humana, siendo esta de calidad la parte referente al desarrollo farmacéutico del medicamento. Hay bastantes similitudes entre los dos tipos de medicamentos (de hecho, el grupo que evalúa la calidad de la EMA es común a veterinaria y humana), debiéndose especificar en esta parte del expediente de registro de un medicamento veterinario lo siguiente: descripción del producto, descripción del método de fabricación, producción y control de materias primas (una parte abierta incluida en el dossier y otra cerrada, del proveedor de materias primas), los controles intermedios y el control de producto terminado (incluyendo estabilidad). Las únicas diferencias relativas a la calidad suelen ser las formas farmacéuticas específicas veterinarias, que no existen en humana y se definirán más adelante.
La Parte 3, de seguridad, se corresponde con el módulo 4 de humana, y se subdivide específicamente en otras dos partes. Una parte A) referente a la toxicología, en la que se realizan las pruebas y estudios comunes de farmacología y toxicología –compartidas con los medicamentos de uso humano– en animales de experimentación en laboratorio, pero no en las especies de destino. En esa parte A) hay requerimientos adicionales concretos para los medicamentos veterinarios, como son los estudios sobre la capacidad de inducir resistencias y otros estudios especiales, que vienen dados por la capacidad de producir alimentos de algunas especies.
Los estudios sobre resistencias se dividen en dos grandes bloques: uno referido a animales productores de alimentos y un segundo referido a animales no productores de alimentos. Para los animales productores de alimentos el VICH3 dictó una Directriz específica –Directriz GL27–, que recoge que la identificación del riesgo se enfoca al potencial de desarrollo de resistencias de bacterias zoonóticas y comensales (que afectan tanto a animales como a seres humanos), identificación del azar (probabilidad de que se produzca, probabilidad de exposición del hombre a ese azar, contacto directo con alimentos, etc.) y las consecuencias para la salud. En el caso de los animales de compañía es más o menos igual, pero para la estimación no se tiene en cuenta la exposición por alimentos, sino solo por contacto directo.
En el caso de los medicamentos veterinarios no solo se evalúa la seguridad del “paciente”, sino que se investigará, además del riesgo para los animales, el riesgo para los humanos (seguridad para el usuario y consumidor) y el riesgo para el medioambiente, dos partes diferentes al dossier de humana. La seguridad para el usuario debe tener en cuenta a la persona que administra el medicamento (veterinario, granjero, responsable del animal) y a la persona que va a estar en contacto con los animales (por ejemplo, niños). La evaluación del riesgo medioambiental (ERA) se hace en 2 fases: una primera en que se valora el grado de exposición alto o bajo y, solo si se supera un umbral de exposición definido en distintas directrices, se pasa a una segunda fase en que se hacen estudios de ecotoxicidad específicos y una evaluación final del riesgo, buscando medidas que lo mitiguen o determinando el riesgo-beneficio del medicamento.
La parte B), de establecimiento de tiempos de espera, es en la que se hace un estudio de depleción de residuos, se fija un residuo marcador para ver cuándo su nivel decae por debajo del límite máximo de residuos (LMR) pre-establecido, y se define un método analítico para determinar este residuo marcador y poder establecer el tiempo de espera que garantice la seguridad para el consumidor. A este respecto, se define como LMR la cantidad máxima de residuos del medicamento que se puede alcanzar en los tejidos del animal –alimento– para considerar que su ingesta es inocua para el consumidor; se fija ese valor para sustancias con actividad farmacológica (el principio activo, pero también metabolitos o excipientes), en distintos tejidos (carne, leche, huevos), y por especies, para una indicación y dosis concretas. Hay casos en que no es necesaria la fijación de ese LMR.
Una vez fijado el LMR por la Comisión Europea, los solicitantes de autorización deben justificar ante la autoridad competente el tiempo de espera, definido como el periodo que hay que respetar entre la administración del medicamento al animal y su sacrificio para la obtención de carne u otro alimento (leche, huevos, miel, vísceras, etc.), asegurando que el animal ha metabolizado el medicamento y sus residuos están por debajo de los LMR. El tiempo de espera se fija para cada producto/medicamento veterinario (no para la sustancia activa, puesto que está afectado por la forma farmacéutica), para todas las especies de destino para las que se indica el medicamento, a las dosis mayores recomendadas, y en todos los tejidos necesarios en función de las especies de destino. Aunque no sea necesaria la fijación de un LMR, puede ser que el tiempo de espera no sea 0 (por ejemplo, para hormonas se deja 1 o 2 días). La AEMPS es el organismo garante de que se establezca un tiempo de espera para cada uno de los medicamentos veterinarios destinados a usarse en animales productores de alimentos en España, de modo que se certifique la seguridad de esos alimentos.
La Parte 4 del expediente es la relativa a la eficacia, y es la más diferente respecto a los medicamentos de uso humano: en veterinaria se establece una parte específica de evaluación de eficacia mediante estudios preclínicos y clínicos de farmacocinética y farmacodinamia en cada una de las distintas especies de destino. Se evalúa aquí el desarrollo de resistencias y los riesgos en animales, los estudios de determinación y confirmación de dosis (normalmente se investigan 3 niveles de dosis: la que se considera adecuada, la mitad y el doble), y finalmente se evalúa la tolerancia en las especies de destino, definiendo un margen de seguridad con dosis mayores de las recomendadas (2, 3 o 5 veces) en un periodo de hasta 6 meses en cada una de las especies de destino. Como en humana, se deben aportar los resultados tanto de estudios preclínicos como clínicos (estos últimos se regirán por las buenas prácticas clínicas); los ensayos se deben hacer en las condiciones normales de cría de animales (por ejemplo, en granjas comerciales), es decir, que se pretende que se valore en situaciones reales.
Finalmente, en la Parte 5, de evaluación beneficio/riesgo, se tiene en cuenta el nivel de eficacia, pero también si hay otros medicamentos en el mercado, y otras mejoras terapéuticas que comporte el medicamento, las cuales se pueden considerar beneficio indirecto, como por ejemplo la facilidad de administración. Para que se proceda con la aprobación y registro del medicamento, la suma de riesgos (toxicológicos, para las especies de destino, el consumidor y el medio ambiente) debe ser menor que el beneficio que aporta.
Cabe destacar que para los medicamentos veterinarios también se dispone, como en humana, de distintos tipos de registro:
- Registro nacional: el expediente del medicamento se presenta a la autoridad competente en un estado miembro (en España, la AEMPS) y solo se puede comercializar en ese estado.
- Reconocimiento mutuo: el medicamento debe estar ya autorizado en un estado miembro (de referencia) por el trámite nacional y, posteriormente, este estado pide el reconocimiento del registro a otros estados miembros.
- Procedimiento centralizado: la solicitud se dirige directamente a la EMA. Hay medicamentos que obligatoriamente deben acudir a registro centralizado, como es el caso de los biológicos, terapias avanzadas, etc. Para el resto, el solicitante puede optar por esta opción, que le da la opción –no obligación– de comercializarlo en todos los estados miembros. Estos expedientes los evalúa el Committee for Medicinal Products for Veterinary Use (CMPV) que eleva una propuesta, recayendo la resolución final sobre la Comisión Europea.
- Procedimiento descentralizado: un medicamento no registrado en ningún estado solicita a uno que actúe como estado miembro de referencia, para que haga un primer informe de evaluación, que suele incluir un listado de preguntas al solicitante, y se envía al resto de estados involucrados en el procedimiento, los cuales pueden incorporar más preguntas o matizar el primer informe. El informe final se consensúa y se remite al solicitante para que responda a las preguntas planteadas. El ponente evalúa la respuesta y emite un informe final, con el que los demás pueden o no estar de acuerdo; en caso de desacuerdo puede retirarse del procedimiento el estado discordante, o bien plantear un procedimiento de arbitraje, o a nivel CVMP de la EMA se hace una propuesta final que el estado que no estaba de acuerdo debe admitir.
Tras la autorización por parte de la AEMPS de un medicamento veterinario, este se inscribirá de oficio en el Registro de Medicamentos, asignándoles un número de registro y su correspondiente código nacional, siendo visibles en CIMAvet, el Centro de Información online de Medicamentos Veterinarios de la AEMPS (disponible en: https://cimavet.aemps.es/cimavet/publico/home.html). Cada número de registro se referirá únicamente a una composición, una forma farmacéutica y una dosis por unidad de administración, incluyendo todos los formatos, teniendo asignado cada número de registro una ficha técnica y un prospecto, accesibles desde CIMAvet. Cada uno de los formatos o presentaciones de un medicamento será identificado por su correspondiente código nacional (Muñoz, 2022a).
Por último, algunos tipos de medicamentos veterinarios tienen que cumplir una serie de condiciones particulares para su comercialización:
- Medicamentos homeopáticos sin indicación terapéutica: el etiquetado del embalaje exterior y, en su caso, el prospecto, se ajustarán a las disposiciones generales relativas al etiquetado y prospecto del capítulo II, sección 4 del Reglamento (UE) 2019/6, y deberán identificarse con la leyenda «Medicamento veterinario homeopático».
- Medicamentos destinados a determinadas especies animales que se posean exclusivamente como animales de compañía (de alguna de las siguientes especies: peces de acuario o estanque, peces ornamentales, pájaros de jaula, palomas mensajeras, animales de terrario, pequeños roedores, hurones y conejos): el etiquetado y, en su caso, su prospecto, se ajustarán a las disposiciones relativas al etiquetado y prospecto establecidas en los artículos 10 a 15 del Reglamento (UE) 2019/6.
- Gases medicinales: en su etiquetado, además de lo indicado en el Reglamento (UE) 2019/6, deberán constar los siguientes datos: logotipo o símbolo identificador de los gases medicinales, especificaciones técnicas que deben cumplir, precauciones de suministro y transporte, y otra información adicional de utilidad a solicitud de la AEMPS.
Particularidades: formas farmacéuticas y vías de administración
Quizás la principal particularidad de los medicamentos veterinarios es que estos deben ser administrados por el veterinario o bajo su supervisión, explicando en este segundo caso al responsable del animal cómo realizarla para que sea correcta, esto es, segura y eficaz. La administración de un medicamento veterinario tiene una serie de especificidades:
- El cumplimiento de la posología adecuada es difícil de garantizar en la administración grupal, por lo que deben tenerse en cuenta una serie de consideraciones para procurarla.
- Los tamaños de los envases del medicamento veterinario son muy diferentes, por lo que se debe disponer tanto de envases para un único animal, como de envases para administración a grupos.
- La composición del medicamento es importante, pero también la de los elementos que le van a servir de forma farmacéutica para su administración (piensos, agua, etc.).
- Debe asegurarse que no existe sobredosificación o infradosificación (sobre todo, con antimicrobianos).
- En la administración a colectividades, debe prevenirse la administración no intencionada a los animales no objetivo (por ejemplo, a los perros de una explotación ganadera).
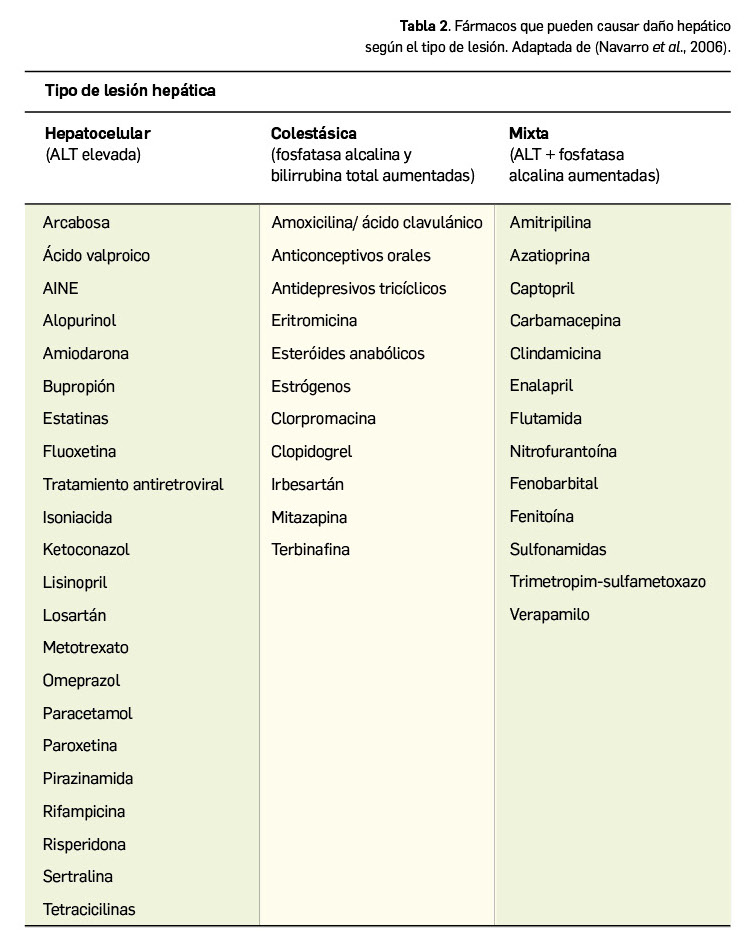
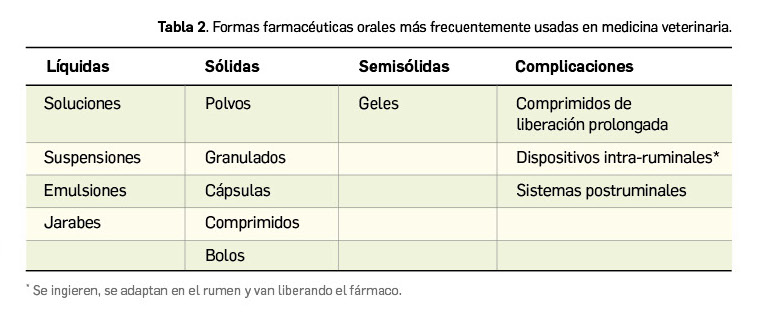
Una característica diferencial relevante de los medicamentos veterinarios, en comparación con los de uso humano, son las distintas formas farmacéuticas. Es preciso subrayar que en la medicina veterinaria existen todas las formas farmacéuticas y vías de administración usadas en humanos, pero, además, existen algunas formas específicas para los animales (Tabla 2). Todas ellas se incluyen dentro de la vía de administración oral, que es la más frecuente en veterinaria, por tener ventajas respecto a otras vías de administración: además de no ser invasiva, permite tratar tanto a animales de forma individual como a grupos de animales. A diferencia de lo que ocurre en humanos, en veterinaria hay que tener en cuenta la especie, la categoría y el tamaño del animal, así como los distintos tipos de producción ganadera (por ejemplo, ganado porcino vs. ganado ovino), ya que el diseño de las granjas es diferente.
La vía oral para uso individual se utiliza principalmente en animales de compañía y se considera de bajo riesgo en cuanto a errores de dosificación. Los medicamentos están disponibles en un amplio rango de concentraciones que permitan tratar a todo el rango de animales en función de su peso, ya que no es lo mismo, por ejemplo, tratar a un perro adulto de 3 kg que a uno de 40 kg.
Además, algunos animales tienen dificultad para ingerir los medicamentos. Para abordar ese problema, en el caso de los perros se puede enmascarar la medicación con porciones de comida. En cambio, los gatos son más difíciles de tratar, por lo que se han desarrollado formulaciones específicas, como los medicamentos palatables a los que se adicionan aromas, edulcorantes u otras sustancias, a fin de que el animal lo ingiera fácilmente. Por su parte, los dispositivos intra-ruminales permiten en animales rumiantes que, una vez ingerido, este se almacene en el rumen liberando el medicamento, para ser posteriormente degradado por los ácidos o expulsado por el animal.
El uso de la vía oral para administración grupal puede ser más problemático, y se encuentran más requisitos específicos a la hora de administrar los medicamentos. Se dispone en la actualidad de tres grupos de alternativas, que se definen a continuación.
- Administración de medicamentos en el pienso. Esta opción puede abordarse mediante:
- Premezclas medicamentosas: se preparan para elaborar piensos medicados o productos intermedios destinados a fábricas de pienso pequeñas, a fin de conseguir una mejor homogeneidad. El pienso medicado debe estar prescrito por un veterinario, debe ser el alimento principal durante todo el tratamiento y debe elaborarse de forma que se asegure que contiene la dosis correcta para el tratamiento previsto (por número de animal y nº de días necesarios). Para ello, hay dos directrices específicas que plantean los requerimientos de calidad que deben tener las premezclas medicinales, una de la EMA [Requisitos de Calidad adicionales para los productos destinados a ser incorporados a la alimentación animal – EMEA/CVMP/080/95-FINAL, de julio de 1997,] y otra del VICH [Directriz GL8 CVMP/VICH/836/99-Final, sobre las Pruebas de Estabilidad para premezclas], que recogen las condiciones para incorporar la premezcla al pienso medicamentoso a fin de asegurar que este es homogéneo, compatible con otros potenciales integrantes del pienso, y cumple las condiciones de estabilidad.
- El pienso medicamentoso resultante debe ser homogéneo con el fin de que cada animal reciba la dosis correcta, y debe tenerse en cuenta la ración que come cada animal, considerando que puede existir competencia entre los animales y que los enfermos comen menos. Para asegurar la administración de la dosis correcta, la legislación establece que el pienso medicamentoso debe corresponderse al menos con el 50% de la ración diaria de pienso; así, al incorporar la premezcla al pienso, su concentración no debe ser inferior al 0,5% del total del pienso medicamentoso. Debe tenerse en cuenta el tipo de pienso, considerar tanto la forma de la premezcla como la del pienso (seco, líquido, pellets), analizando las posibles interacciones entre los componentes del pienso y los de la premezcla. También hay que determinar una vida útil, la estabilidad tanto para la premezcla como para esta una vez combinada con el pienso, aportando a los usuarios recomendaciones con respecto al procesamiento y compatibilidad, y a las condiciones necesarias para la fabricación del pienso (sobre todo, en cuanto a humedad y temperatura), para garantizar la integridad de la premezcla.
- Los piensos medicamentosos exigen para su administración instalaciones adecuadas: debe disponerse de 2 silos por nave (uno para pienso normal y otro para el medicamentoso) con el fin de evitar la contaminación cruzada. Los silos deben estar limpios en su parte externa y en su parte interna y, tras agotarse el pienso medicamentoso, se debe limpiar el silo que los contenía para evitar la contaminación del pienso que se incorpore posteriormente. Además, los silos deben estar cerrados para evitar el acceso de pájaros o roedores, la lluvia o contaminantes arrastrados por el viento.
- Top-dressing: es una forma específica de administración a través del pienso, mediante la cual el medicamento veterinario se esparce sobre la superficie del alimento, inmediatamente antes de alimentar a los animales. Se puede utilizar a nivel individual o colectivo para un número reducido de animales. Se hace sobre una pequeña cantidad de alimento primero para asegurar que el animal ingiere la ración completa y, por tanto, la dosis correcta. Sin embargo, es difícil asegurar que llegue la dosis correcta a todos los animales, ya que no se garantiza la homogeneidad en la distribución del medicamento, por lo que la incertidumbre de alcanzar la dosis correcta es mayor que con las premezclas. La palatabilidad puede favorecer el rechazo del pienso, lo que también dificulta alcanzar la dosis adecuada. Este tipo de administración apenas se usa en España, si bien en otros países –como Alemania– se emplea mucho en las granjas.
- Pienso líquido: se obtiene mezclando pienso seco, agua y el medicamento veterinario. Se prepara en la granja, normalmente mezclando primero el medicamento veterinario con una pequeña cantidad de pienso y agregando agua hasta el límite necesario. Esta forma de administración permite administrar vacunas (reconstituida previamente en el solvente proporcionado, luego mezclándola con el alimento y, finalmente, con el agua). Al igual que las premezclas, este tipo de mezclas deben cumplir unas condiciones de administración: la tasa de incorporación y la homogeneidad deben ser adecuadas, la calidad del agua debe ser buena (igual que para la administración de medicamentos en agua), y debe asegurarse la compatibilidad con otros ingredientes del pienso y la estabilidad del pienso líquido.
2. Administración de medicamentos en agua o leche. Puede hacerse con 2 tipos de formas farmacéuticas:
- Polvos orales: son una mezcla del fármaco con otros excipientes que se administran como polvo o disuelto en agua/leche.
- Soluciones orales: sistema disperso y homogéneo consistente en la mezcla de ≥ 2 componentes para administrar directamente o disuelto en agua/leche. Dentro de estas, quizá la solución más usada es la leche medicada, que se administra en aquellos animales recién nacidos que aún se alimentan con leche materna. El ganadero la prepara poco antes de la administración y se administra a una temperatura de 37-40ºC. Pueden usarse sustitutos de la leche, que deben disolverse en agua caliente a > 70ºC; en estos casos, teniendo en cuenta que muchos principios activos pueden degradarse a alta temperatura, debe dejarse enfriar antes de añadir el medicamento para su posterior administración. Es importante valorar la solubilidad del medicamento en la leche a una cierta temperatura y añadirlo poco a poco para garantizar su disolución y la homogeneidad de la mezcla. Además, hay que tener igualmente en cuenta las incompatibilidades por interacciones; por ejemplo, nunca administrar tetraciclinas en la leche, pues la formación de complejos con los cationes Ca2+ reduce su absorción.
Una serie de consideraciones generales deben ser tenidas en cuenta para asegurar la correcta administración de medicamentos en agua/leche: i) solubilidad e hidrosolubilidad: el medicamento debe ser soluble en el líquido en cuestión o, en caso contrario, se deben usar excipientes que aumenten su solubilidad (por ejemplo, reguladores del pH); ii) calidad4: estabilidad química y microbiológica, para lo cual se añaden conservantes y antioxidantes; iii) la velocidad de disolución debe ser lo suficientemente alta para disolver la dosis en poco tiempo; y iv) las características organolépticas específicas: si el sabor es desagradable deben añadirse excipientes para facilitar que los animales lo tomen (por ejemplo, edulcorantes frente al sabor amargo del paracetamol), o puede colorearse el agua intencionadamente para diferenciar el agua medicada de la no medicada (algunos medicamentos colorean el agua por sí mismos), evitando que sea rechazada por los animales. En base a lo anterior, se debe considerar el consumo medio de agua diario de los animales a tratar, la homogeneidad del agua medicada, la probabilidad de acceso de los animales al suministro de esa agua (que no exista un suministro alternativo de agua para que se asegure que beben el agua medicada), así como la incompatibilidad del agua con otros elementos.
- Bloques para lamer que incorporan medicamentos: se usan poco actualmente porque dificultan la dosificación, empleándose fundamentalmente para administrar complejos vitamínicos y minerales y otros complementos de la dieta.
En definitiva, si bien hay casos específicos (Tabla 3), es imprescindible conocer las características del medicamento mediante la información del producto (ficha técnica) con el fin de seleccionar el medio más adecuado para su administración oral y, una vez elegido, contemplar las condiciones de administración que se han descrito en cada uno de ellos para lograr que la administración sea segura tanto para el animal como para el medio ambiente (Muñoz, 2022b).

Condiciones de dispensación
La legislación vigente establece que los medicamentos veterinarios o de uso animal pueden ser dispensados en tres tipos de establecimientos: farmacias comunitarias, comerciales veterinarias detallistas con servicio farmacéutico garante, y entidades y agrupaciones ganaderas que cuenten con servicio farmacéutico garante y solo para sus socios. No obstante, la ley autoriza a la distribución y venta por otros establecimientos, en los términos previstos, exclusivamente de medicamentos veterinarios sin receta destinados a perros, gatos, animales de terrario, pájaros domiciliarios, peces de acuario y pequeños roedores.
Es preciso diferenciar, como así lo hace el artículo 15 del RD 1157/2021, entre dos tipos de medicamentos veterinarios según la necesidad o no de receta, que es, como en medicamentos de uso humano, una decisión competencia de la AEMPS establecida en base a criterios científicos.
- Medicamentos no sujetos a prescripción veterinaria. En este grupo se encuentran medicamentos con efectos antisépticos y desinfectantes tópicos, antihelmínticos y ectoparasiticidas; también se englobarían otros productos que no tienen consideración de medicamento. Los requisitos que se deben cumplir para proceder a la dispensación son: por un lado, seleccionar un medicamento que está autorizado en la indicación requerida y para la especie en cuestión (determinantes de eficacia y de la seguridad) y explicar al responsable del animal la forma de administrarlo y la pauta a seguir para un uso seguro del mismo.
- Medicamentos sujetos a prescripción veterinaria. Son la mayoría de los medicamentos comercializados para su uso en animales, incluyendo las fórmulas magistrales y preparados oficinales, que solamente podrán ser preparadas y dispensadas en las oficinas de farmacia autorizadas. Se incluirán todos aquellos medicamentos que solo puedan ser administrados por el veterinario o los servicios veterinarios oficiales, o bajo su control o supervisión.
El artículo 37 del RDL 1/2015, que recoge el texto refundido de la Ley de garantías y uso racional de medicamentos y productos sanitarios, establece la exigencia de prescripción para aquellos medicamentos que requieran la adopción de medidas especiales para evitar riesgos ya sea para la especie de destino, la persona que los administra, otros animales o el medio ambiente. También se exige prescripción a los medicamentos destinados a tratamientos o procesos patológicos que requieran un diagnóstico preciso previo por un veterinario, a los medicamentos con sustancias psicoactivas (estupefacientes o psicótropos), los destinados a animales productores de alimentos (con las excepciones que legalmente se establezcan), los utilizados en los supuestos de prescripción excepcional por vacío terapéutico –incluidos los preparados oficinales, fórmulas magistrales y autovacunas–, los gases medicinales, los medicamentos que contengan un principio activo que lleve menos de 5 años autorizado y los medicamentos inmunológicos. También se exigirá la receta veterinaria para la dispensación de cualquier medicamento veterinario prescrito por veterinarios de otro Estado miembro de la UE, no establecidos en el territorio nacional y que presten sus servicios en España, y para todos aquellos medicamentos de uso humano que se vaya a emplear en animales.
En el suministro de medicamentos a veterinarios debe considerarse que cualquier veterinario colegiado podrá retirar en la farmacia comunitaria los medicamentos que solicite para su botiquín, siempre que sean de uso veterinario (a excepción de anestésicos locales para odontología). La farmacia emitirá una factura en la que conste el nombre del veterinario, número de colegiado y dirección, las cantidades de medicamentos suministrados con número de lote y caducidad de cada uno de ellos; guardará una copia firmada y sellada durante 5 años, a disposición de la inspección.
La dispensación excepcional de medicamentos de uso hospitalario a clínicas veterinarias o veterinarios se contempla en el Real Decreto 1132/2010, de 10 de septiembre, que establece que el suministro de estos productos se realiza por la farmacia previa petición del veterinario de los medicamentos precisos mediante hoja de pedido. Estos medicamentos están destinados al ejercicio de su actividad profesional y para el uso o administración por él mismo. En la hoja de pedido deberán figurar: la identificación del veterinario y de su colegiación, denominación del medicamento y número de ejemplares, fecha y firma. La farmacia sellará y firmará el documento, indicando la fecha de la dispensación y lo entregará al veterinario, quedándose con una fotocopia de la hoja de pedido como justificante de dicho suministro.
La receta veterinaria
Con la excepción de las recetas de estupefacientes, la prescripción veterinaria carece de un formato homogéneo. No se ha llegado por ahora a un consenso para disponer de una única plataforma de receta electrónica veterinaria, conviviendo varias de ellas, lo cual supone una desventaja frente a la dispensación de medicamentos de uso humano. Sin embargo, bien sea en formato papel o electrónico, están definidos legalmente5 los datos mínimos que debe incluir para que se considere válida y pueda procederse a su dispensación. La receta veterinaria tendrá validez en todo el territorio nacional y se editará en la lengua española oficial del Estado y en las respectivas lenguas cooficiales en las Comunidades Autónomas que dispongan de ella.
Los datos mínimos que tiene que contener una receta para proceder a su dispensación son:
- datos identificativos del veterinario prescriptor: nombre y dos apellidos, dirección completa, número de colegiado y provincia de colegiación;
- denominación del medicamento perfectamente legible, especificando la forma farmacéutica, el formato (si existen varios) y el número de ejemplares a dispensar;
- firma del prescriptor;
- fecha de la prescripción;
- si la receta es para animales de abasto, deberá contar con los siguientes datos adicionales: código de identificación de la explotación, tiempo de espera (indicado aun cuando sea de 0 días) y número de receta.
El veterinario puede hacer constar por escrito en la receta, o documento aparte, las instrucciones para el propietario o responsable de los animales sobre el uso o administración del medicamento. La duración del tratamiento y la validez de la receta para su dispensación no será superior a 30 días, salvo en el caso de enfermedades crónicas o tratamiento periódicos (que serán de 3 meses), haciéndolo constar así en la receta. La receta debe contar de 3 ejemplares: una para el veterinario prescriptor, una para el responsable del animal y una para el centro dispensador; tanto el prescriptor como el centro dispensador deben archivar su copia durante 5 años a disposición de las autoridades. No se ha establecido legalmente un número máximo de envases que pueden dispensarse en una receta, limitándose la cantidad prescrita y dispensada al mínimo necesario para el tratamiento del animal, según el criterio del veterinario, que deberá tener en cuenta los diferentes formatos de medicamentos veterinarios existentes que se adecúen al tratamiento prescrito.
En el caso de que no se disponga del medicamento recetado por el veterinario, y no sea posible la sustitución por el veterinario, el farmacéutico –exclusivamente– podrá proceder a su sustitución por otro con distinto nombre comercial, pero con la misma composición cualitativa y cuantitativa de principio/s activo/s, forma farmacéutica, vía de administración, dosis y registro para la especie animal a la que va destinado el medicamento, informando al propietario y con su consentimiento. El farmacéutico indicará en la parte posterior de la receta normalizada, el nombre del medicamento dispensado y firmará, junto con el propietario o responsable del animal o grupo de animales, en la zona indicada para ello. En el caso de medicamentos prescritos a animales destinados a consumo, además de cumplir las con-diciones anteriores, únicamente podrán sustituirse por un medicamento que, según su ficha técnica, tenga un tiempo de espera igual o inferior al medicamento originalmente prescrito, manteniéndose el tiempo de espera especificado por el veterinario. No son susceptibles de sustitución, por el contrario, aquellos medicamentos veterinarios inmunológicos, o aquellos que, por razón de sus características de biodisponibilidad y estrecho rango terapéutico, así lo determine el Ministerio de Sanidad de acuerdo con el Ministerio de Agricultura, Pesca y Alimentación.
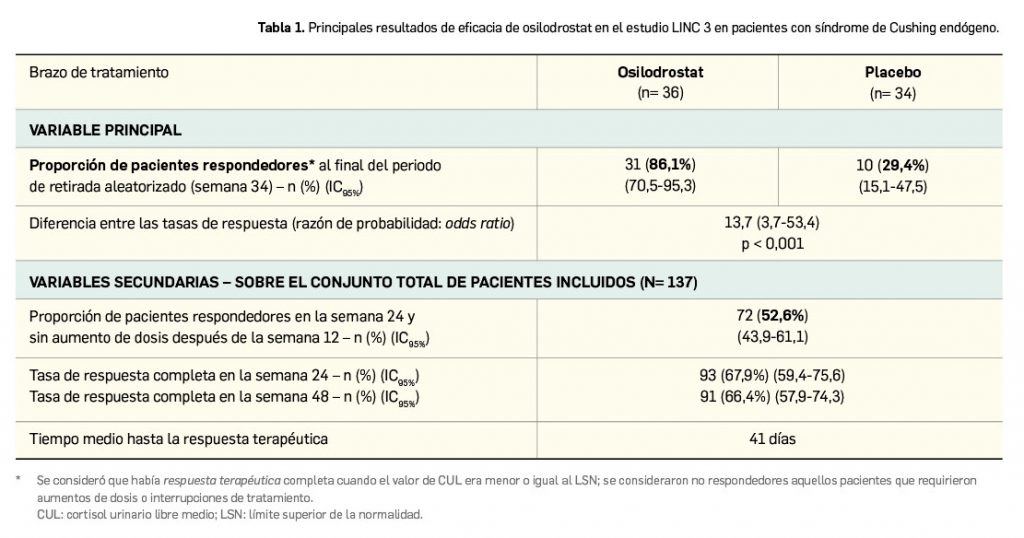
Por otro lado, cuando no existe un medicamento indicado para tratar una enfermedad en una especie concreta, el veterinario procederá a hacer una prescripción excepcional por vacío terapéutico, recurriendo a un medicamento que considere, bajo su responsabilidad, que puede curar la patología a tratar y evitar sufrimientos innecesarios a los animales, indicándolo en la receta correspondiente. Puede producirse en este caso lo que se conoce como “prescripción en cascada”, con un protocolo de niveles definido, como si se tratase de los peldaños de una escalera, que implica que, si existe un medicamento autorizado en un nivel, no se puede saltar al siguiente:
- Prescribirá en primer lugar un medicamento veterinario registrado en España para esa especie animal, pero para otra enfermedad, o un medicamento veterinario con efecto terapéutico similar al deseado para esa misma enfermedad, aunque con registro para otra especie animal.
- Un medicamento de uso humano autorizado en España o un medicamento veterinario con similar efecto terapéutico al deseado autorizado en otro Estado de la UE para esa misma especie u otras y para esa enfermedad u otras. Este escalón puede variar ligeramente en las especies animales productoras de alimento (Figura 2).
- Un medicamento veterinario de fabricación extemporánea, esto es, una fórmula magistral, un preparado oficinal o una autovacuna.

En la prescripción excepcional para animales de abasto, el veterinario fijará un tiempo de espera que considere adecuado en el caso de que no figure para esa especie, no siendo inferior a lo establecido por la Comisión Europea. Además, las recetas de fórmulas magistrales o preparados oficinales deberán incluir, junto a los datos generales descritos anteriormente, los siguientes: composición cualitativa y cuantitativa, proceso patológico a tratar, especie animal y cantidad.
Por su parte, las recetas de estupefacientes están reguladas en el capítulo 3 del RD 1675/2012 (publicado en el BOE del 29 de diciembre de 2012), modificado por la Orden PRE/2436/2013, y se ajustarán al modelo establecido (Figura 3). Suelen contar con medidas de seguridad que eviten la falsificación. La prescripción será para un solo animal y de un solo medicamento, especialmente destinada a la dispensación de medicamentos que contengan sustancias estupefacientes de la Lista I de la Convención Única de 1961 sobre Estupefacientes (no de las listas II y III de dicha convención) y las que a nivel nacional sean consideradas reglamentariamente como tales. La duración del tratamiento no superará los 30 días y la validez de la receta es de 10 días.
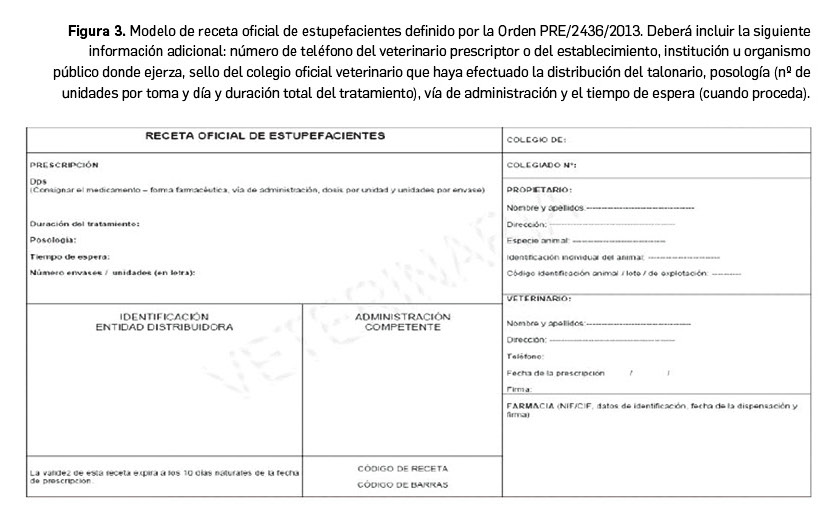
En la dispensación de estupefacientes, debe verificarse la validez de la receta y la copia de la misma se archivará durante 5 años. Las oficinas de farmacia, igual que se hace con los estupefacientes de uso humano, deben hacer una declaración anual de estupefacientes, cada mes de enero, indicando su uso veterinario, que se remite a la Comunidad Autónoma correspondiente o bien directamente a la AEMPS por vía telemática (a través de la aplicación Labofar, en aquellas CCAA que la tienen habilitada). El suministro de estupefacientes a establecimientos veterinarios legalmente autorizados solo podrá efectuarse a aquellos vinculados a la oficina de farmacia autorizada de la misma provincia, y solamente por esta, debiendo cumplir estos establecimientos una serie de requisitos que son establecidos por las Comunidades Autónomas si desean contar con un depósito especial de medicamentos estupefacientes de uso animal (Amado, 2022).
Antibióticos y el proyecto ESVAC
Los antibióticos son uno de los grupos de medicamentos más ampliamente usados en medicina veterinaria, y pertenecen a las mismas clases de fármacos usados en medicina humana. Además de haber salvado millones de vidas e impulsado el desarrollo de la medicina moderna y de la humanidad a través del control de un gran número de patologías bacterianas, como la tuberculosis, desde su descubrimiento –el mayor hito biomédico de la historia6 – y desarrollo han permitido la producción de alimentos y proteínas animales de alta calidad para gran parte de la población mundial. Pero desde el inicio de su uso generalizado en la década de 1940, el ser humano se percató de la facilidad con la que las bacterias se hacían resistentes a los antibióticos cuando se hacía un uso inadecuado de los mismos7. Y es que los mecanismos de resistencias han existido siempre (por ejemplo, las bacterias ambientales que producen antibióticos poseen mecanismos de resistencia intrínsecos), por la gran capacidad de adaptación –mutaciones– y de transmitir el material genético a sus descendentes que poseen las bacterias.
El uso masivo de los agentes antibacterianos, realizado tanto en medicina humana como en veterinaria durante décadas, ha hecho que el problema de las resistencias bacterianas y la selección positiva y diseminación de cepas multirresistentes (capaces de sobrevivir a los efectos de varios antibióticos) se haya convertido en uno de los mayores retos de la salud pública en el presente y en el futuro próximo, por su gran impacto clínico, epidemiológico y microbiológico. Algunas estimaciones han apuntado a que, sin políticas que le hagan frente de forma efectiva, las resistencias a antimicrobianos serán la causa hacia el año 2050 de hasta 10 millones de fallecimientos anuales, superando al cáncer como primera causa de muerte; en la actualidad ya se producen en los hospitales españoles unas 4.000 muertes cada año por infecciones por bacterias multirresistentes.
No obstante, existe una concienciación creciente frente a esta problemática, que puede frenar su avance. Diversas instituciones, como el Centro Europeo para la Prevención y Control de Enfermedades (ECDC), la Autoridad Europea para la Seguridad Alimentaria (EFSA) y la EMA, han publicado informes sobre el análisis del consumo de antibióticos y las resistencias en bacterias patógenas en salud humana y sanidad animal. Los resultados sugieren que, desde la perspectiva One Health o Una sola salud, existe un gran potencial en ambos sectores para desarrollar aún más el uso prudente de los antibióticos, pues están directamente interrelacionados: el uso de antibióticos veterinarios influye en el desarrollo de resistencias antimicrobianas en humanos y viceversa.
A partir de esos informes, es larga la lista de foros y organismos internacionales que trabajan de manera coordinada para afrontar el problema de las resistencias a antibióticos. Así, además de la UE, organizaciones internacionales como la OMS, la FAO, la OIE y el Codex Alimentarius están colaborando desde diferentes puntos de vista, pero complementarios, para definir estrategias comunes y así conseguir resultados de una forma más eficaz.
A nivel nacional, sobresale la actividad que desde su aprobación en 2014 –por el Consejo Interterritorial del Sistema Nacional de la Salud y la Conferencia Intersectorial de Agricultura–desarrolla el Plan Nacional frente a la Resistencia a los Antibióticos (PRAN), con un enfoque integral que contempla salud humana, sanidad animal y medio ambiente. El PRAN ha contado desde su inicio con la colaboración de todas las comunidades autónomas, 8 ministerios (Sanidad, Agricultura, Economía, Educación, Ciencia, Interior, Defensa y Transición Ecológica), 70 sociedades científicas, organizaciones colegiales, asociaciones profesionales y universidades y más de 300 colaboradores expertos. Todos ellos trabajan en permanente contacto bajo la coordinación de la AEMPS.
Los logros alcanzados por el trabajo de esta red de expertos en cada una de las líneas estratégicas, a pesar de la crisis sanitaria que ha supuesto la COVID-19, constituyen avances muy significativos en la estrategia común de preservar de manera sostenible la efectividad de los antibióticos existentes. Entre otros, se ha mejorado el sistema de vigilancia del consumo de antibióticos, se han aprobado distintos programas de uso prudente de antibióticos (PROAs) en medicina humana y animal, y se han consensuado los indicadores comunes para la vigilancia del consumo y resistencias. Según los datos divulgados en 2021, el consumo de antibióticos en salud humana en España había mostrado un 32% de reducción respecto al periodo 2014-2020, que ha sido mayor incluso en sanidad animal: se ha registrado una reducción del 56% en las ventas de antibióticos de uso veterinario respecto a ese mismo periodo, pasando de 402 mg/PCU8 en 2015 a 172 mg/PCU en 2019. Actualmente, el PRAN sigue avanzando en su tarea orientada a reducir el consumo, y la necesidad de consumo, de antibióticos en medicina humana y veterinaria.
Conviene subrayar que existe un listado de antibióticos de especial importancia en la medicina humana que se denominan antibióticos de importancia crítica (CIA), sobre los que existe un amplio consenso en torno a la necesidad de usarlos con suma precaución en medicina veterinaria (Figura 4). La resistencia a los antibióticos en animales puede propagarse al ser humano no únicamente a través del contacto con las mascotas domésticas, sino también a través de los alimentos, del agua y otras vías de contaminación ambiental: por ello resulta imprescindible la monitorización de las bacterias patógenas, zoonóticas y comensales que permita identificar patrones emergentes de resistencias.

En definitiva, la vigilancia del consumo de antibióticos de uso veterinario es imprescindible. Debe estar basada en una red amplia que proporcione una fuente de datos fiable y representativa del consumo real, que cubra desde los datos de ventas de antibióticos por los laboratorios farmacéuticos hasta los niveles de comercialización más cercanos al consumidor final, así como las prescripciones veterinarias y el uso de antibióticos en granjas. Basado en estos tres pilares, se puede alcanzar un conocimiento completo de la situación real de consumo de antibióticos en animales de un país, aportando información de calidad desde una perspectiva One Health para la toma de decisiones políticas que impulsen las medidas de reducción de forma multisectorial.
En la línea de promoción del uso prudente de antibióticos (esto es, un uso racional y específico que maximice su efecto terapéutico y minimice el desarrollo de resistencias), merece mención aparte el proyecto ESVAC (European Surveillance of Veterinary Antimicrobial Consumption), que constituye la base del sistema de vigilancia nacional del consumo de antibióticos veterinarios. Se trata de un proyecto de ámbito europeo, coordinado por la EMA, de recogida, validación, análisis y evaluación de datos sobre la venta y el consumo de medicamentos veterinarios que contienen en su composición antibióticos como principio activo. Los resultados, obtenidos en la unidad estandarizada a nivel de la UE (mg/PCU), se comunican anualmente con referencia al ejercicio de compraventa del año anterior al vigente.
Los datos brutos de ventas en ESVAC se obtienen mediante la declaración de los laboratorios, los almacenes mayoristas, las oficinas de farmacia, los establecimientos comerciales detallistas y las entidades o agrupaciones ganaderas autorizadas para la dispensación de medicamentos veterinarios. Estos tres últimos están obligados legalmente9 a proporcionar los datos que se les solicitan, mientras que los laboratorios los aportan con carácter voluntario. Los datos se cargan mediante una aplicación web (https://sinaem.aemps.es/ESVAC) y se refieren al número de unidades vendidas de cada formato de medicamento en concreto en el año previo. Actualmente participan de forma voluntaria 31 países europeos. El periodo de notificación comienza habitualmente a principios de año, y se prolonga hasta aproximadamente mediados de año.
El último informe ESVAC10, publicado en noviembre de 2021, refleja que en la UE el volumen de ventas de antimicrobianos para su uso en animales productores de alimentos se ha reducido en un 43% entre 2011 y 2021. De particular relevancia se considera que en ese periodo se haya reducido en un 13% la venta y uso de fluoroquinolonas, en un 33% para las cefalosporinas de 3ª y 4ª generación, en un 77% para polimixinas y en un 85% para otras quinolonas. La situación en los 25 países que reportan datos tiene contrastes sustanciales: si bien 19 países muestran un descenso de las ventas de antimicrobianos veterinarios de > 5%, 2 países solo reportan un descenso pequeño (< 5%) e incluso 4 países notifican un aumento de su uso superior al 5%. La reducción de más del 50% en el uso de estos medicamentos en algunos países en ese periodo refleja el vasto potencial de reducir su consumo por parte de otros países.
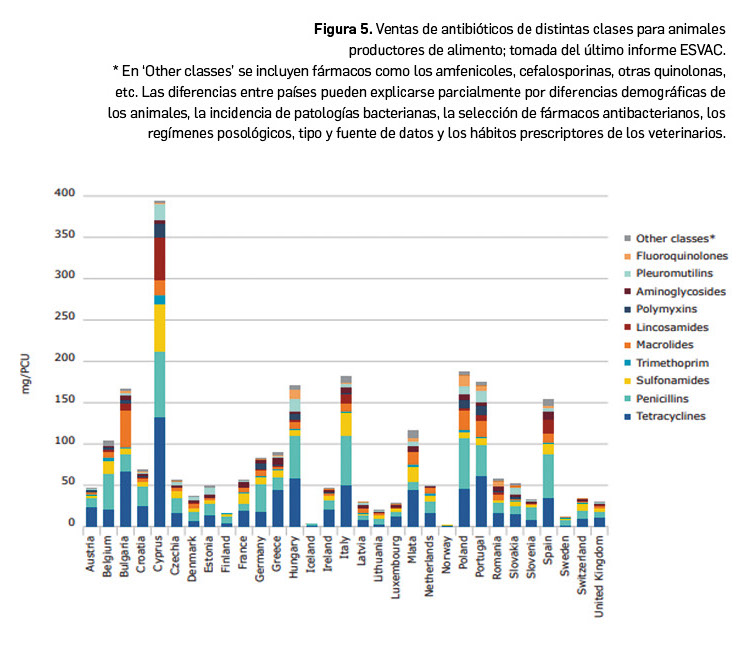
Por países (Figura 5), España se sitúa destacada a la cabeza de todos los países de Europa en cuanto al número total de toneladas de antimicrobianos vendidas (1.246, de un total de 5.577 entre los 31 países que notifican). No obstante, cuando se ajusta por población de animales, cae al séptimo puesto de la lista, con 154,3 mg/PCU, pero por encima de la media del conjunto de países europeos (89,0 mg/PCU). Los datos para nuestro país incluidos en ESVAC se corresponden con los citados en el PRAN, con un notable descenso, respecto a 2014 (año en que se inició el PRAN), de un 70% en 2019 y un 63% en 2020. La mayor reducción se ha producido en las ventas por parte de los laboratorios (-59% entre 2014 y 2019) y, en cuanto a fármacos, el descenso más llamativo se dio en el uso de polimixinas (-98% hasta 2020). En los últimos años esa tendencia favorable se ha mantenido, evidenciándose un descenso de un 45% y un 33%, respectivamente, frente a las ventas de antibióticos en 2017. Los grupos de antibióticos más usados en España en 2020 son penicilinas, tetraciclinas, lincosamidas y sulfonamidas, todas ellas clasificadas en los grupos B y C, para los que se acepta su uso con cautela o con prudencia. Por especies, más del 75% de las prescripciones de antibióticos se hacen en cerdos, un 14% en aves de corral y un 7% en ganado vacuno.
De forma complementaria al proyecto ESVAC, otras dos iniciativas impulsadas en nuestro país pueden contribuir a la vigilancia de la resistencia a antibióticos. Así, el primero de ellos es el programa PresVet, que entró en vigor en enero de 2019, a raíz de la publicación el 17 de abril de 2018 del Real Decreto 191/2018, de 6 de abril, por el que se establece la transmisión electrónica de datos de las prescripciones veterinarias de antibióticos destinados a animales productores de alimentos para consumo humano, y se modifican diversos Reales Decretos en materia de ganadería. Este nuevo RD reguló las condiciones y requisitos aplicables a la transmisión electrónica de estas prescripciones, y estableció que los veterinarios deben proporcionar a la Administración los datos relativos a los antibióticos que prescriban a animales en las explotaciones en las que trabajan.
Por otra parte, en el marco del PRAN, se han puesto en marcha una serie de programas llamados “Programas Reduce”, para la reducción voluntaria de determinados antibióticos en diferentes especies animales como ovino y caprino, así como en porcino, cunicultura, bovino de carne y leche, avicultura de carne (pollos broiler) y animales de compañía. Estos programas se basan en que siempre, antes de prescribir un antibiótico, se deben tener en cuenta las recomendaciones generales de uso responsable, guías de prescripción y categorización de antibióticos críticos en veterinaria a efectos de limitar su uso a los casos estrictamente necesarios (Sacristán, 2022).
Farmacovigilancia de medicamentos veterinarios
La farmacovigilancia veterinaria atiende tanto a los riesgos que el medicamento pueda suponer para los animales a los que se administra y a los que puedan entrar en contacto con ellos, como a los riesgos para las personas (que administren el medicamento, estén en contacto con los animales o consuman productos derivados de estos)
o para el medio ambiente.
Tiene una base legal y práctica de más de 40 años en la UE, periodo durante el que se ha sometido a vigilancia, desde la perspectiva de la eficacia y la seguridad, a los medicamentos veterinarios. En el Reglamento (UE) 2019/6 se define la farmacovigilancia veterinaria como “la ciencia y las actividades vinculadas a la detección, la evaluación, la comprensión y la prevención de las sospechas de acontecimientos adversos o cualquier otro problema relacionado con un medicamento”. Esto es, un sistema posautorización que controle la eficacia y la seguridad de los medicamentos veterinarios en condiciones reales de uso, cuando los medicamentos se emplean en distintas condiciones de manejo, en animales de todas las edades, con patologías múltiples y tratamientos complejos, a veces en especies animales que no están entre las autorizadas del fármaco, etc. Arroja una información muy útil de cara a conocer en profundidad las bondades y los riesgos que entraña el uso de medicamentos veterinarios.
Hay que recordar que la farmacovigilancia se sustenta fundamentalmente en la notificación espontánea de las Sospechas de Acontecimientos Adversos (SAA) por parte de los profesionales sanitarios (obligatoria, según se recoge en el RDL 1/2015), sean veterinarios o farmacéuticos, ya que son quienes diagnostican, prescriben, dispensan y, muchas veces, administran los medicamentos, por lo que por sus conocimientos técnicos y por la cercanía al animal enfermo están en la mejor disposición para identificar y valorar la presentación de SAA. También notifican SAA los laboratorios de la industria farmacéutica. Se deben notificar todas las SAA, con independencia de su gravedad, de si están descritas o no en ficha técnica, o de cualquier otra consideración.
En resumidas cuentas, como en el caso de los medicamentos de uso humano, la detección de un riesgo asociado a un medicamento veterinario desemboca en la adopción de medidas reguladoras por parte de la AEMPS: la modificación de las condiciones de autorización, la suspensión temporal, la retirada de un lote, la exigencia de un estudio poscomercialización o la suspensión definitiva.
Cuando surge una alerta con un medicamento veterinario, la AEMPS la pone en conocimiento de todos los agentes implicados en el sector, para proceder a la retirada del mercado de dichos lotes y evitar efectos adversos por su uso. En la farmacia comunitaria se actuará procediendo a la inmovilización inmediata de los lotes afectados y a la devolución, por los cauces reglamentarios, a los proveedores.
Un nivel de participación insuficiente por parte de los profesionales sanitarios redundará en una menor cantidad y calidad de información a los sistemas de farmacovigilancia que se verán en dificultades para poder aumentar el conocimiento de los medicamentos veterinarios provocando que las conclusiones y, en su caso, la implementación de medidas de gestión de los riesgos se retrase y llegue demasiado tarde. Por consiguiente, es imprescindible aumentar el número de notificaciones de SAA a los sistemas de farmacovigilancia y, aunque obvio, es esencial que la calidad de la información aportada en las notificaciones sea la mayor posible, pues de esa manera se consigue una mejor comprensión de acontecimientos adversos y puede haber una mejor y más acertada gestión de los riesgos.
Conviene aclarar que la farmacovigilancia veterinaria no es un sistema de control del ejercicio profesional, es decir, que no se enjuicia la corrección o idoneidad de la instauración de un tratamiento o de su administración, sino que lo que se busca es conocer el comportamiento del medicamento y ver si hay nuevos riesgos o riesgos conocidos pero que tienen una mayor gravedad que aconsejen la adopción de medidas reguladoras para controlarlos. De igual manera, no es un sistema de resolución de conflictos entre laboratorios, profesionales sanitarios y ganaderos o propietarios de los animales que quieran reclamar unas compensaciones por unos daños hipotéticamente causados por un medicamento (que deberían canalizarse por otras vías distintas).
El nuevo Reglamento (UE) 2019/6 ha introducido numerosos cambios en este campo, enfocados a llevar a cabo una farmacovigilancia más basada en los riesgos y tratando de eliminar o disminuir la carga burocrática. Sobresalen los siguientes:
- La simplificación de los métodos de notificación por parte de los titulares de autorizaciones de comercialización. Todas las SAA, con independencia del país donde ocurran, deben notificarse a la base de datos de farmacovigilancia veterinaria europea (EVVET 3).
- Simplificación de los plazos en que los titulares deben notificar las SAA, que pasan a ser de 30 días, sin distinguir entre tipos de SAA como ocurre ahora.
- Se añaden las reacciones adversas en animales a medicamentos de uso humano al ámbito tradicional de aplicación (reacciones adversas en animales y en personas, faltas de eficacia, insuficiencias de los tiempos de espera, efectos nocivos en el medio ambiente y transmisión de agentes infecciosos debidos a los medicamentos veterinarios).
- Desaparición de los Informes Periódicos de Seguridad (IPS) que los titulares debían enviar periódicamente a las autoridades competentes. Estos IPS se sustituyen por la nueva obligación de realizar con periodicidad un proceso de gestión con todos sus medicamentos que traten de identificar las señales de seguridad con la mayor antelación posible.
La notificación de una SAA puede hacerla el profesional sanitario directamente a la AEMPS o también al titular de la autorización de comercialización del medicamento sospechoso de haberla causado. Puede hacerse empleando el formulario existente a los efectos (la Tarjeta Verde, editada por la AEMPS) o en cualquier otro soporte (por ejemplo, electrónicamente a la base de datos nacional de farmacovigilancia veterinaria FV VIGÍA-VET, por fax o por correo electrónico), pero lo importante es que contenga la información necesaria.
Desde que en 2008 se creara el Sistema Español de Farmacovigilancia de Medicamentos Veterinarios (mediante el RD 1246/2008), el nivel de notificación de SAA con el uso de medicamentos veterinarios en España ha crecido notablemente en los últimos años, superando los 1.500 casos, sin duda debido al éxito de los programas de promoción. Sin embargo, sigue existiendo una infranotificación importante, ya que, considerando las poblaciones animales y el volumen de tratamientos instaurados anualmente, sería esperable recibir unas 3.000 a 3.500 notificaciones individuales, por lo que las campañas de promoción que están en marcha desde hace más de una década (cursos, jornadas y publicaciones) siguen siendo necesarias para elevar el nivel de conocimiento y concienciación de los profesionales sanitarios respecto de sus obligaciones legales en materia de farmacovigilancia veterinaria (Casimiro, 2022).
Los resultados del Boletín Anual sobre farmacovigilancia veterinaria recientemente divulgado (AEMPS, 2022) recogen un total de 2.139 notificaciones de acontecimientos adversos (AA) en España durante el año 2021; de ellas, 1.594 fueron casos notificados por primera vez (en la línea de años precedentes: 1.510 notificaciones en 2020 y 1.579 en 2019), y el resto, eran seguimientos que aportaban una mayor información sobre casos notificados previamente. Estos AA se notificaron mayoritariamente en perros (1.039 eventos), seguido de las siguientes especies animales: gato (193), oveja (101), cerdo (72), vaca (70), conejo (37), aves de corral (20), cabra (9), caballo (6), hurón y visón. En cuanto al tipo de medicamentos con que sea asociaron estos AA, sobresalen las vacunas, por delante de los ectoparasiticidas (Figura 6). No obstante, de todos ellos solo 25 AA afectaron a personas.
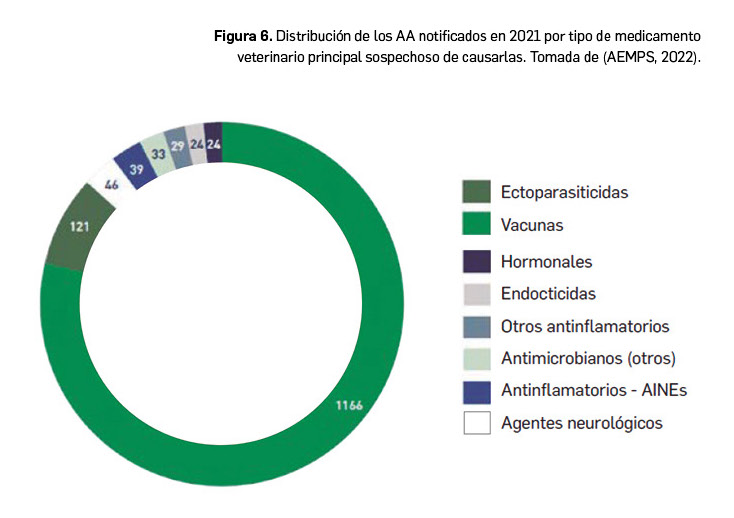
Respecto del origen de las notificaciones iniciales, en 2021 de nuevo fue mayoritaria la realizada por los titulares de autorización de comercialización de los medicamentos o TAC (1.387 AA, un 88%), frente al resto de posibles orígenes –profesionales sanitarios, de la ganadería, propietarios de animales, otros usuarios, etc.–, que fueron responsables de la notificación del restante 12% (189 AA).
La AEMPS recibió en 2021 un total de 1.636 IPS relativos a medicamentos veterinarios, solo alguno menos que en el año 2020 (1.700), manteniéndose en cifras similares a la última década. En el marco del Worksharing, un programa aprobado por la red de Jefes de Agencias (MHA) para distribuir la responsabilidad de evaluación de los IPS entre países, se recibieron un total de 319 IPS, habiendo actuado España como evaluador principal responsable de 14 moléculas o combinaciones: amoxicilina+ácido clavulánico+prednisolona, flumethrin+propoxur, moxidectina oral, cloprostenol, melarsomina, ciclosporina, cefapirina+prednisolona, ácido tolfenámico y oxitetraciclina. En 31 de los IPS, el Comité de Seguridad de Medicamentos Veterinarios, como órgano colegiado de la AEMPS para el asesoramiento técnico y científico en lo relativo a farmacovigilancia veterinaria, concluyó que era necesario proceder a la modificación de las fichas técnicas de los medicamentos.
Pese a todo lo expuesto, en 2021 no se emitió ninguna alerta por razones de farmacovigilancia de medicamentos veterinarios. Sí se recibieron y gestionaron, por afectar a medicamentos registrados en España, 6 informaciones de seguridad no urgentes (NUIS) por razones de farmacovigilancia. En la función de velar por la calidad de los medicamentos, la AEMPS emitió 4 alertas por defectos de calidad.
Panorama actual
A mitad de junio de 2022, los datos disponibles en CIMAvet reflejan que el arsenal de medicamentos veterinarios en España cuenta con un total de 10.125 presentaciones autorizadas, de las cuales 2.228 no están comercializadas de forma efectiva. La forma farmacéutica mayoritaria son los inyectables parenterales y los comprimidos de administración por vía oral, que totalizan prácticamente 3.700 y 2.300 presentaciones, respectivamente (37% y 23% del total de autorizados). Es preciso recordar que todos los medicamentos veterinarios tienen un código nacional superior a 570000 e inferior a 600000 (a partir de ese número ya son medicamentos de uso humano).
Según cifras de Veterindustria11 (Asociación Empresarial Española de la Industria de Sanidad y Nutrición Animal), la agrupación de los laboratorios que investigan y fabrican medicamentos veterinarios, la comercialización de este tipo de medicamentos orientados a la sanidad y nutrición animal mueve anualmente en España una cantidad de dinero que ilustra su relevancia en el sistema productivo de nuestro país, con un amplio impacto económico adicional al sociosanitario. Así, se mantiene en tendencia ascendente, habiéndose registrado un crecimiento global en 2021 del 9,3%, hasta alcanzar los 1.877 millones de euros; de ellos, 1.187 millones corresponden al mercado nacional (variación de +8,6% respecto al año previo) y 690 millones a las exportaciones (+10,4%), que suponen casi el 37% del mercado global gracias a que las empresas del sector zoosanitario español –29 de ellas asociadas en Veterindustria– están presentes en más de 90 países de todo el mundo. La mayor parte de la producción industrial se orienta a los productos farmacológicos (47%) y biológicos (28%), para su uso en tres grupos de especies mayoritarios: perros y gatos (32%), ganado porcino (28%) y ganado vacuno (21%).
Las estimaciones del sector sugieren que, sin los productos para la salud y la nutrición animal, los ganaderos europeos necesitarían criar un 89% más de vacas, un 54% más de cerdos y un 25% más de aves para mantener los actuales niveles de producción de alimentos; por tanto, al mejorar la salud y el rendimiento del ganado, disminuye la presión ejercida por la agricultura sobre la naturaleza. La vigilancia, prevención y control de las enfermedades animales, que se ven favorecidos por el uso racional de los medicamentos veterinarios, permitirán afrontar el reto de alimentar entre 2050 y 2060 con alimentos seguros y de calidad a una población mundial que se espera supere los 9.800 millones de personas; esto será posible si se cuenta con animales de producción sanos y explotaciones ganaderas en condiciones higiénico-sanitarias adecuadas, al mismo tiempo que se protegen los diversos ecosistemas en una producción agropecuaria sostenible.
La evolución histórica de la innovación terapéutica en veterinaria permite vislumbrar un futuro prometedor (Figura 7). Desde que los nuevos antibióticos impulsaran el crecimiento en las décadas de los años 50 y 60 del siglo pasado, algunos de los hitos más relevantes fueron la aparición de los productos antiparasitarios que permitieron la expansión de los animales productores de alimentos o el desarrollo durante la década de 1980 de los primeros endocidas y las vacunas producidas por biotecnología; ese avance benefició también de manera directa a la medicina humana, pues las primeras vacunas utilizadas en ingeniería genética fueron desarrolladas por una empresa de la industria de sanidad animal. Un ejemplo claro de cómo la industria de productos veterinarios puede adaptarse al desarrollo de medicamentos de uso humano ha sido la investigación clínica de una vacuna frente a la COVID-19 por parte de la empresa española Hipra. No obstante, la norma general es que el periodo que transcurre desde que se descubre un medicamento hasta que se fabrica y distribuye pasan una media de 8-10 años, un periodo ligeramente menor que para los medicamentos de uso humano, pero para nada desdeñable.
Por otra parte, conviene subrayar que la EMA trabaja desde varios ángulos para favorecer el desarrollo de la innovación en medicamentos veterinarios y la promoción de la salud animal y salud pública en Europa, codo con codo con las empresas implicadas en el desarrollo y comercialización de esos medicamentos. Además de contar con el ya citado CVMP, en 2015 creó ad hoc el Grupo de Expertos en Terapias Innovadoras en Veterinaria (grupo ADVENT), para aportar un consejo científico útil sobre los requerimientos de autorización de productos innovadores en este campo.
Dicha agencia ha publicado recientemente el informe anual correspondiente al año 2021 (EMA, 2022), que recalca los logros más importantes, también en medicina veterinaria. Destaca que recomendó la autorización de comercialización de 12 nuevos medicamentos veterinarios que han incluido 7 nuevos principios activos (frente a los 92 nuevos medicamentos y 54 principios activos en humana), la mayoría de ellos para su uso en perros (5), gatos (3) y cerdos (2). De los 12 nuevos medicamentos, 5 eran vacunas y, de ellas, 4 se producen por biotecnología. Sobresale por su grado de innovación terapéutica una nueva vacuna para cerdos que tiene el potencial de reducir la necesidad de tratamiento antimicrobiano y podría limitar el desarrollo de resistencias bacterianas: se trata de un medicamento para la inmunización pasiva de los lechones recién nacidos (mediante la inmunización activa de las cerdas reproductoras), indicada frente a las consecuencias clínicas de la infección por Clostridioides difficile y Clostridium perfringens.
Además de informar sobre los datos de ventas de antimicrobianos veterinarios, el informe recoge que las ventas de antibióticos considerados de importancia crítica para su uso en humanos se redujeron notablemente entre 2011 y 2020, representando solo el 6% de las ventas totales. En términos de seguridad, el sistema EudraVigilance recogió la notificación de más de 80.000 reacciones adversas a medicamentos de uso animal, más de 55.000 en perros y más de 12.000 en gatos. El año pasado, la EMA también se preparó para la implementación del nuevo Reglamento de medicamentos veterinarios y para el lanzamiento del nuevo Sistema de Información de Ensayos Clínicos que respalda la puesta en marcha del Reglamento de Ensayos Clínicos, que entró en vigor en enero de 2022.
El papel asistencial del farmacéutico
Si bien el perfil profesional del farmacéutico se ha ido especializando en distintos campos, el conocimiento de los fármacos es la esencia de la profesión en todos ellos. Por tanto, como profesional sanitario experto en la materia, tiene un papel fundamental en toda la vida de los medicamentos veterinarios: como medicamentos que son, están sujeto a estrictas medidas de control técnico y jurídico, desde la investigación hasta los procesos de fabricación, comercialización, distribución y dispensación.
En todo el proceso, el farmacéutico se erige como una figura imprescindible. Puede participar en el nivel de la investigación básica, desempeñándose en el mundo académico, con capacitación para la identificación y aislamiento de principios activos de distintas fuentes, así como en la síntesis química, la química analítica, la biotecnología y los estudios farmacológicos a diferentes escalas. También desde el ámbito de la farmacia industrial y galénica, los farmacéuticos contribuyen al desarrollo y mejora de formas farmacéuticas, optimizando los procesos de obtención que posibilitan la incorporación al arsenal terapéutico de muchos fármacos eficaces in vitro, la consecución de mejores niveles de eficacia o la mejora de su tolerabilidad, todo ello enfocado a mejorar los resultados en salud de la farmacoterapia. En investigación clínica, hay farmacéuticos especialistas que a menudo son figuras relevantes en el diseño y desarrollo de estudios impulsados por la industria, en tareas diversas como las relacionadas con la gestión de los medicamentos (recepción, custodia y preparación), el registro de los movimientos del producto en investigación (entrada, dispensación, devolución o gestión de restos/residuos), etc.
Los farmacéuticos también contribuyen a la salud pública por la vía de la distribución de los medicamentos veterinarios, y aseguran la calidad alimentaria mediante su desempeño como profesionales públicos en los controles sanitarios sobre la presencia de residuos en animales y alimentos de origen animal. No obstante, habida cuenta de las citadas limitaciones normativas de dispensación de los medicamentos veterinarios, que no están financiados por el Sistema Nacional de Salud, serán los farmacéuticos comunitarios y aquellos que ejerzan como garantes en comerciales veterinarias detallistas y entidades y agrupaciones ganaderas, los que mayor potencial de actuación tengan en pro de la salud animal y el uso optimizado de estos medicamentos.
Si bien en los últimos tiempos los medicamentos veterinarios han tenido limitada visibilidad en la farmacia comunitaria, la oportuna promoción del enfoque One Health los ha vuelto a sacar a la palestra por su relevancia sanitaria, en especial desde la mayor concienciación derivada de la irrupción de la COVID-19 o el brote de monkeypox12 (viruela del mono). Con su integración efectiva en el primer nivel asistencial del SNS, la Farmacia adquiere gran importancia en la lucha contra el impacto13 de las zoonosis. Se trata del establecimiento sanitario más accesible (sin necesidad de cita previa), ubicuo y cercano, por el que pasan diariamente más 2 millones de personas y desde donde se ofrecen más de 182 millones de consejos sanitarios al año. Cuando no se trata de una situación de urgencia, para el ciudadano es más fácil, cómodo y rápido acudir a una farmacia a retirar el medicamento para su mascota o animales de abasto, pues está abierta durante un amplio horario y con guardias periódicas; todo ello, sin perjuicio de los necesarios controles veterinarios que cada animal requiera. De ahí que la red española de más de 22.000 farmacias comunitarias en la que trabajan más de 55.000 farmacéuticos resulte especialmente interesante como centro de divulgación para transmitir una información científicamente rigurosa sobre los medicamentos veterinarios y sus características diferenciales.
Se describen, a continuación, las principales vías asistenciales de actuación del profesional farmacéutico para con los ciudadanos propietarios de mascotas o explotaciones ganaderas, en favor de la salud global.
I. Educación sanitaria orientada a la prevención
El principal mensaje que cualquier profesional sanitario debe transmitir a los ciudadanos será el recordatorio de que las zoonosis, como enfermedades infecciosas transmitidas de animales al hombre, pueden ser de varios tipos –priónicas, bacterianas, víricas, fúngicas y parasitarias–, y la variedad de mecanismos de transmisión hace difícil establecer en muchos casos eficaces protocolos de prevención, que difieren para cada patógeno. Es esencial conocer al menos las que son objeto de vigilancia en el entorno cercano para recomendar las medidas más adecuadas en cada caso.
Ciertas prácticas se consideran eficaces para reducir el riesgo a nivel comunitario y personal: directrices para el cuidado de los animales en el sector agrícola que ayudan a reducir la posibilidad de brotes zoonóticos de origen alimentario a través de alimentos como la carne, los huevos, los productos lácteos o incluso algunas verduras; cumplimiento de las normas relativas al agua potable y a la eliminación de residuos; la protección de las aguas superficiales en el medio natural; o la promoción del lavado de manos después del contacto con animales. Otras medidas específicas son útiles frente a la prevención de ciertas zoonosis: la salmonelosis o la toxoplasmosis se pueden evitar con un adecuado lavado y cocinado de los alimentos; las enfermedades transmitidas por vectores (como la leishmaniosis o la enfermedad de Lyme) se pueden prevenir con una protección frente a las picaduras mediante repelentes o mosquiteras, y la protección de la mascota con collares antiparasitarios; la vacunación de humanos o el uso de mascarillas son eficaces frente algunas zoonosis transmitidas por el aire (como la tuberculosis o la COVID-19); y la vacunación de los perros o la evitación del contacto directo con murciélagos pueden ser medidas efectivas frente a la rabia o la fiebre Q, respectivamente.
Este tipo de medidas preventivas, como parte del esfuerzo común realizado entre veterinarios, farmacéuticos, ganaderos, industria farmacéutica y autoridades sanitarias, es lo que permite la obtención de alimentos seguros desde el punto de vista de la ausencia de medicamentos en los mismos. La observancia de la profilaxis será cada vez más importante si se considera que el 75% del entorno terrestre y el 65% del entorno acuático han sido gravemente alterados por los seres humanos, y que el 25% de la cubierta forestal perdida es suficiente para aumentar la probabilidad de contacto entre humanos y ganado con animales silvestres, lo cual puede aumentar la posibilidad de transmisión de zoonosis. En definitiva, dada la interrelación con el medio ambiente, resulta necesaria la concienciación de todos para que se asuman políticas de salud pública que favorezcan la construcción de sistemas de producción animal más sostenibles y tomen en cuenta los factores que aumentan el riesgo y dificultan el control de las zoonosis, tales como el cambio climático, los incendios forestales y la deforestación, los viajes intercontinentales, el abandono de animales en la vía pública o la mutación de los agentes etiológicos con otros genotipos.
Las últimas encuestas14 realizadas al respecto muestran un creciente conocimiento en la población europea sobre los beneficios de los medicamentos veterinarios. Así, por ejemplo, en torno a 2 de cada 3 personas cree que los medicamentos veterinarios tienen un impacto positivo en el bienestar de los animales de granja y que la vacunación de estos ayuda a prevenir la transmisión de enfermedades a las personas. En relación a los animales de compañía, hay una mayor concienciación sobre la necesidad de profilaxis, constatándose que el 76% de los encuestados está de acuerdo con la vacunación regular de mascotas, el 78% en visitar con regularidad (al menos 1 vez al año) al veterinario, y el 80% cree que es importante la prevención de pulgas y garrapatas. Pese a todo, alrededor del 60% de los ciudadanos siente que no está debidamente informado sobre los medicamentos veterinarios y su uso, el 59-62% no sabe que el uso de hormonas o de antibióticos veterinarios como promotores del crecimiento no está permitido en la UE, o el 54% no sabe que para garantizar la seguridad de los alimentos se debe respetar un tiempo desde el uso del medicamento veterinario hasta el sacrificio del animal.
Por todo ello, la instrucción que desde la farmacia se puede ejercer sobre los usuarios respecto a los medicamentos veterinarios tiene un gran potencial educativo. En esa línea resulta interesante subrayar el impacto que sobre la salud global tiene la vacunación de animales: detener la enfermedad en su origen ayuda a evitar consecuencias devastadoras para el bienestar animal, a reducir las pérdidas de animales y de alimentos, de modo que protege el planeta y evita graves consecuencias para la salud pública. Otro aspecto esencial en el enfoque One Health es la concienciación sobre el uso racional de los antibióticos, orientado a minimizar su consumo lo máximo posible para combatir la propagación de las resistencias bacterianas.
Para una mayor información de la población sobre medicamentos veterinarios, es conveniente que conozcan la existencia de CIMAvet (https://cimavet.aemps.es/cimavet/publico/home.html), base de datos de la AEMPS donde se puede encontrar un buscador avanzado que permite buscar y filtrar por: nombre del medicamento, número de registro o código nacional, especie de destino, forma farmacéutica, principios activos, estado de comercialización, condiciones de dispensación, o ciertas características específicas (si es psicótropo, estupefaciente o antibiótico). Desde esa propia página, se puede acceder al Nomenclátor Veterinario, una herramienta que proporciona información básica sobre los medicamentos para la prescripción electrónica y también de consulta a los profesionales sanitarios y administraciones competentes en materia de medicamentos veterinarios. Dicho Nomenclátor contiene todos los medicamentos veterinarios comercializados, así como los suspendidos en los 12 meses previos y aquellos anulados en el plazo de 6 meses anteriores, incluyendo los datos para su identificación y una información sobre sus características técnicas.
Cabe recordar que la venta a distancia a través de páginas web está regulada por el Real Decreto 544/2016, de 25 de noviembre, por el que se regula la venta a distancia al público de medicamentos veterinarios no sujetos a prescripción veterinaria. Son solo esos medicamentos sin receta los que pueden venderse (no así los que la requieran, incluidas fórmulas magistrales, preparados oficinales, y autovacunas), solo desde farmacias y establecimientos comerciales detallistas que hayan notificado esta actividad a la autoridad competente de la comunidad autónoma, y siempre con la intervención de un farmacéutico responsable, que facilitará el asesoramiento personalizado al usuario. En la venta no pueden participar intermediarios, a excepción de la empresa encargada del transporte hasta la vivienda del destinatario. Además, la farmacia podrá aplicar un descuento sobre el precio de venta al público de hasta un 10%, pero no podrá ofrecer regalos, premios, obsequios, concursos, bonificaciones o actividades similares como medios vinculados a la venta a distancia. El destinatario final de la venta debe encontrarse en la UE, y en caso de ser de un país diferente a España, el medicamento en cuestión deberá estar autorizado en aquel país, respetándose su normativa en relación a esta modalidad de venta.
Para una mayor información sobre los requerimientos de los sitios web que pueden llevar a cabo la venta online de medicamentos veterinarios y de la forma de proceder ante pedidos de los mismos se recomienda consultar la guía editada por el Consejo General de Colegios Farmacéuticos (CGCF, 2022).
II. Optimización de la farmacoterapia
Desde el punto de vista de los resultados en salud (animal) con el uso de medicamentos veterinarios, los servicios profesionales que el farmacéutico puede desarrollar son similares a los relativos a medicamentos de uso humano, con algunas particularidades. Así, una vez establecido el diagnóstico y prescrito un tratamiento por el veterinario, el farmacéutico debe velar por su uso racional para que permitan a los pacientes alcanzar el máximo beneficio clínico. En líneas generales, las instrucciones dadas por el médico deben ir siempre reforzadas con recomendaciones relativas a las medidas preventivas por parte del farmacéutico.
A un ciudadano que acuda a la farmacia comunitaria por un problema de salud de su mascota, se le puede ofrecer –a través del Servicio de Indicación–, si no existe contraindicación, un medicamento no sujeto a receta veterinaria e indicado para el abordaje de síntomas menores de la especie en cuestión. Si ese problema no mejorara en un corto periodo de tiempo, se debe aconsejar al dueño que acuda a consulta con un veterinario. Para conocer si un medicamento está sujeto a prescripción veterinaria o no, se puede consultar CIMAvet, donde se identifican los medicamentos con los mensajes de “Sujeto a prescripción veterinaria” o “No sujeto a prescripción veterinaria”.
En el momento de la dispensación de cualquier medicamento frente a una patología animal, el farmacéutico comprobará que el dueño cuenta con toda la información necesaria para que su uso sea efectivo y seguro. Se debe averiguar si existe algún criterio que impida la dispensación, tal como alergia a algún componente del medicamento, una contraindicación absoluta o interacciones con otros medicamentos (o alimentos), entre otros. Si es la primera vez que el animal va a recibir un medicamento, la labor se centrará en asegurar que el propietario sale de la farmacia conociendo para qué es y cuál es su correcto proceso de uso. En una dispensación de continuación, el farmacéutico evaluará si el beneficio-riesgo del medicamento sigue siendo favorable, verificando si ha habido cambios en el tratamiento (dosis, pauta, duración, adición de nuevos medicamentos, etc.) o si el animal ha experimentado algún problema de seguridad que pudiera hacer sospechar de una reacción adversa, interacción, contraindicación, etc. Es, además, muy importante, evaluar y promocionar la adherencia al tratamiento prescrito durante el proceso terapéutico, como uno de los pilares de la labor asistencial farmacéutica.
En el caso de medicamentos sujetos a receta, la dispensación debe ir precedida obligatoriamente por la comprobación de su validez y autenticidad. Para salvar el inconveniente de que no haya una sola plataforma de receta electrónica, en caso de necesidad, puede solicitarse al prescriptor que remita a la farmacia por e-mail la receta cumplimentada y firmada electrónicamente para poder imprimirla y dispensarla de igual forma que si se tratase de una receta en papel. La dispensación deberá realizarse en los envases originales intactos, salvo que los formatos autorizados del medicamento posibiliten una dispensación fraccionada sin que se vea comprometida la integridad del acondicionamiento primario del medicamento y siempre que vayan acompañados de la documentación preceptiva. El farmacéutico dispensador deberá sellar y fechar la copia de la receta reservada al propietario o responsable, una vez le sea presentada esta y haya procedido a su dispensación, almacenando posteriormente el original de la receta durante 5 años.
Cuando el veterinario proceda a la prescripción excepcional de un medicamento de uso humano para el empleo en un animal, la dispensación de este medicamento se limita a una farmacia legalmente autorizada, siempre que la receta incluya la leyenda de “prescripción excepcional”. En tal caso, se debe hacer una dispensación informada que, posteriormente, debe ser anotada en el libro recetario, no solo cuando se trate de psicótropos o estupefacientes (estos deben ser anotados también en el libro de estupefacientes), si bien en estos dos últimos casos se debe comprobar también la identidad de la persona que retira el medicamento, anotando en la receta veterinaria el número de DNI. No pueden dispensarse al público general –solo a veterinarios– medicamentos de uso humano calificados por la AEMPS como hospitalarios.
Finalmente, un adecuado seguimiento farmacoterapéutico, ofrecido por el farmacéutico de forma rutinaria, sistematizada y registrada/documentada, con reuniones periódicas con el propietario del animal, permitirá maximizar los beneficios en salud de la farmacoterapia, así como detectar, atenuar y resolver la posible aparición de resultados negativos y problemas relacionados con la medicación. La farmacovigilancia ante posibles reacciones adversas, y la identificación y prevención de interacciones farmacológicas y contraindicaciones de los medicamentos revertirá en una mejor calidad de vida de los animales. Con ese objetivo, es fundamental instruir al propietario acerca de las manifestaciones de los efectos adversos más relevantes del tratamiento (y favorecer que los comunique a su veterinario), así como concienciarle sobre la conveniencia de evitar la toma de medicación no recomendada por el veterinario.
___________________________________________________________________________
Para una dispensación informada y un seguimiento farmacoterapéutico óptimo, además de la recomendación de consultar las fichas técnicas de los medicamentos, si se tiene en consideración que la información científica se actualiza constantemente, cobran especial relevancia las bases de datos que contienen información farmacológica actualizada y pormenorizada sobre medicamentos veterinarios. Es el caso de BOT PLUS (disponible en: https://botplusweb.farmaceuticos.com), un aplicativo informático que incluye todos los medicamentos veterinarios registrados por la AEMPS e incluidos en CIMAvet. Para cada presentación se dispone, además de un acceso a su ficha técnica y prospecto oficiales, la siguiente información:
- Información administrativa del medicamento: código nacional, número de registro, nombre completo, laboratorio comercializador, grupo ATC, estado de autorización y comercialización, forma farmacéutica, vía de administración o IVA aplicable. BOT PLUS no informa sobre precios de comercialización de los medicamentos veterinarios, al tratarse de medicamentos de precio libre y no existir ningún precio oficial de aplicación.
- Condiciones de dispensación: BOT PLUS identifica si el medicamento está sujeto a prescripción veterinaria, si es estupefaciente o psicótropo, o si se trata de un medicamento de administración exclusiva por el veterinario o bajo su control.
- Condiciones de conservación: información codificada relativa a si el medicamento es de conservación en nevera, a temperatura inferior a 25ºC o a 30ºC o si se tiene que proteger de la luz. Igualmente existe una serie de mensajes de advertencia para conocer los periodos de caducidad y de validez tras su apertura.
- Composición cualitativa y cuantitativa.
- Indicación del medicamento para las diferentes especies de destino.
- Tiempos de espera.