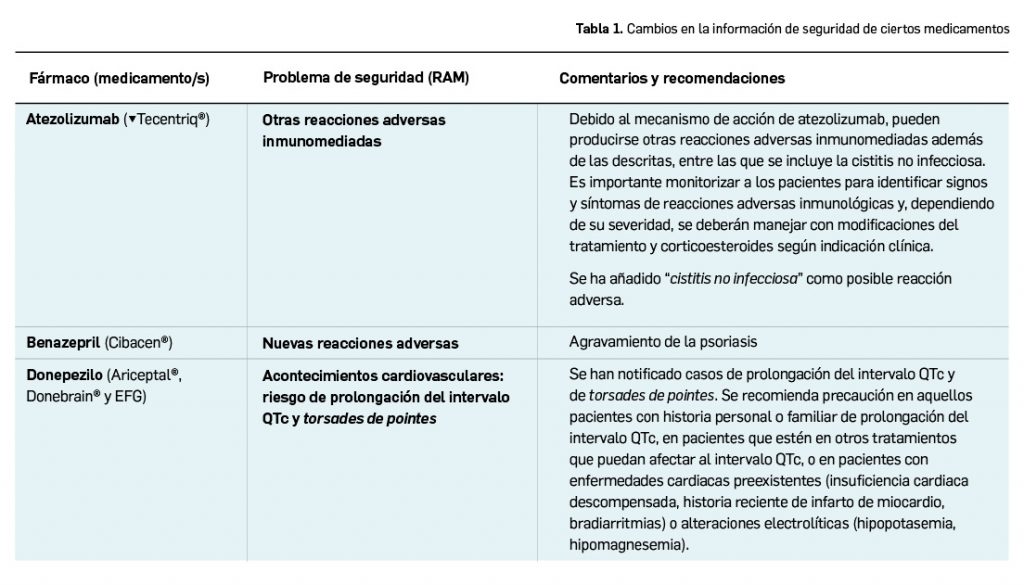
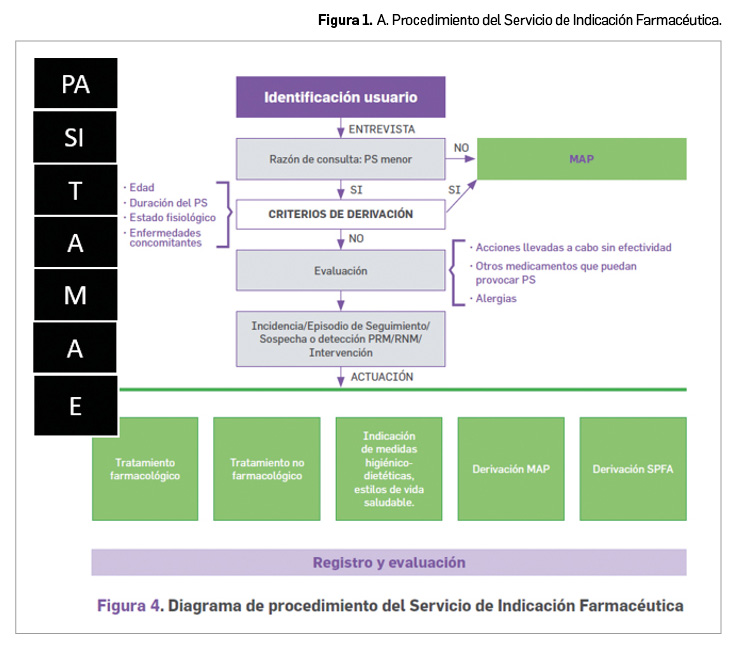
La agencia reguladora de Canadá, Health Canada, ha anunciado nueva información en la ficha técnica de pramipexol, que incluye una advertencia del riesgo de síndrome de abstinencia de agonistas dopaminérgicos (SAAD, o DAWS, por sus siglas en inglés). Se ha iniciado una revisión con el resto de los agonistas dopaminérgicos.
En Canadá, la autoridad reguladora de medicamentos, Health Canada, ha actualizado la monografía del producto (CPM, Canadian Product Monograph) de los medicamentos con pramipexol (Mirapexin®) para incluir una advertencia sobre el riesgo de síndrome de abstinencia de agonistas de la dopamina (SAAD, o DAWS, por sus siglas en inglés). Esta información es similar a la ya existente en la ficha técnica del medicamento en España.
Se debe recordar que los agonistas de la dopamina están indicados para tratar la enfermedad de Parkinson, el síndrome de piernas inquietas y la acromegalia. En Canadá están disponibles los siguientes agonistas dopaminérgicos: apomorfina, bromocriptina, cabergolina, pergolida, pramipexol, quinagolida, ropinirol y rotigotina.
Health Canada ha estado monitorizando el riesgo potencial de SAAD desde 2019, luego de las actualizaciones realizadas por la autoridad reguladora en Japón (PMDA). En 2020, el fabricante de pramipexol actualizó voluntariamente el CPM para incluir una advertencia de SAAD, desencadenando la revisión de seguridad de Health Canada para todos los agonistas de la dopamina comercializados en Canadá. La citada agencia revisó la información de las bases de datos canadienses e internacionales de reacciones adversas notificadas y la literatura científica. Se evaluaron veintitrés (23) notificaciones de casos (dos canadienses y 21 internacionales) de SAAD en pacientes tratados con agonistas dopaminérgicos.
Dicha revisión ha establecido un vínculo entre el uso de los agonistas dopaminérgicos pramipexol, quinagolida o ropinirol y el riesgo de SAAD y, por lo tanto, Health Canada trabajará con los fabricantes de los medicamentos con quinagolida (Norprolac®) y ropinirol (Adartrel®, Requip®, Requip-Prolib®, Rolpryna® y EFG) para actualizar los CPM, a fin de que incluyan una advertencia de SAAD. Estos medicamentos en España ya contienen esa advertencia sobre SAAD, con información tal como “los pacientes deben ser monitorizados atentamente durante la disminución gradual y la interrupción. En caso de síntomas de abstinencia graves y/o persistentes, se puede considerar la readministración temporal de ropinirol a la mínima dosis efectiva”.
Recomendaciones
Health Canada trabajará con los fabricantes de estos agonistas de la dopamina para incluir el riesgo potencial de SAAD/DAWS como precaución en su ficha técnica. Por ahora, no hay suficiente información para establecer un enlace entre el riesgo de SAAD y otros agonistas de la dopamina como la apomorfina (Apo-Go Pen®, Dacepton®), bromocriptina (en Parlodel® ya se advierte), cabergolina (Dostinex®, Sogilen®), pergolida y rotigotina (en Neupro® ya se advierte).