Resumen
Fostemsavir es un nuevo antirretroviral en forma de profármaco que se hidroliza en nuestro organismo por acción de la fosfatasa alcalina en la luz del tracto gastrointestinal a la fracción activa temsavir. Este se une directamente a la subunidad gp120 de la glicoproteína gp160 de la envoltura del VIH-1 y provoca que esa glicoproteína se mantenga en una conformación “cerrada”: es así capaz de inhibir selectivamente la interacción entre el virus y el receptor celular CD4, evitando la entrada de los viriones y la infección de células huésped. En base a ello, y a la probada actividad antiviral de temsavir frente a distintos subtipos del VIH-1 (reducida frente al subtipo AE del grupo M y ausente frente a los grupos O y N), el medicamento ha sido autorizado para el tratamiento oral diario, en combinación con otros antirretrovirales, de adultos infectados por el VIH-1 multirresistente para quienes, de otro modo, no es posible establecer un tratamiento antirretroviral supresor.
Los datos clínicos que sustentan su aprobación de uso adicional a un régimen multifármaco de TAR oral derivan fundamentalmente de la cohorte aleatorizada de un estudio de fase 3 (N= 272 pacientes altamente pretratados y con limitadas opciones terapéuticas) con un brazo de control que se mantuvo únicamente los 8 primeros días de tratamiento. En el día 8, el uso de una monoterapia funcional con fostemsavir demostró una eficacia significativamente superior a placebo (p < 0,0001), evidenciada en una reducción unas 4 veces mayor en la carga de ARN viral en sangre (-0,79 log10 vs. -0,17 log10 con placebo; descenso neto a favor del fármaco de -0,63 log10). A más largo plazo, aunque no se puede comparar con un control, es destacable que el uso de fostemsavir con un régimen optimizado con 1-2 fármacos activos adicionales se asocia con una eficacia mantenida: la tasa de supresión virológica a la semana 96 es del 60%, aumentando desde el 53% a la semana 24. La seguridad del nuevo fármaco parece aceptable y manejable clínicamente, pues las reacciones adversas más comunes (> 10%) durante su uso se refieren a la tolerabilidad gastrointestinal (diarrea, náuseas, dolor abdominal y vómitos), reacciones de hipersensibilidad (erupción cutánea) y cefalea. Entre los eventos considerados como graves (< 4%), durante el seguimiento a largo plazo en el estudio pivotal destacaron la neumonía, la celulitis y los eventos coronarios arteriales. Durante el desarrollo de fostemsavir se registraron 36 muertes, de las cuales una se vinculó con el uso del fármaco, por un caso de síndrome inflamatorio de reconstitución inmune, un evento adverso de especial interés.
A pesar de las dudas sobre el riesgo de aparición de resistencias durante el tratamiento con fostemsavir (los datos apuntan a que podría tener una barrera genética menor que otros fármacos, lo cual podría afectar a la eficacia de un TAR optimizado) y de la necesidad de contar con resultados de eficacia y seguridad en condiciones de vida real, este inaugura una nueva clase de antirretrovirales que puede ser útil como régimen de rescate en pacientes infectados por cepas de VIH-1 multirresistentes. Aunque comparaciones indirectas apuntan a una mayor eficacia de lenacapavir, se debe tener en cuenta que ambos pueden formar parte del mismo régimen, dado que no se han encontrado por ahora resistencias cruzadas con otros fármacos antirretrovirales. Fostemsavir es, por tanto, una opción más dentro del reducido arsenal farmacológico disponible para el tratamiento de las infecciones multirresistentes por VIH-1.
Aspectos fisiopatológicos
El virus de la inmunodeficiencia humana (en adelante, VIH) es un retrovirus o virus de ARN monocatenario retrotranscrito (ssRNA-RT, por sus siglas en inglés) que forma parte de los lentivirus (familia Retroviridae), pese a que su cinética de replicación es muy agresiva y diferente a la forma insidiosa en que se desarrolla la infección por otros lentivirus.
Su genoma se compone de dos copias idénticas de cadenas de ARN que dependen para su replicación en la célula huésped de la enzima denominada transcriptasa inversa (retrotranscriptasa), que da lugar mediante retrotranscripción a un ADN provisional que a menudo se inserta en el genoma del hospedador por la acción de la integrasa viral, una ADN polimerasa dependiente de ARN. El tamaño del genoma del VIH es inferior a 10 000 nucleótidos y contiene varios genes: 3 principales (gag, pol y env) que codifican, cada uno, para varias proteínas (estructurales, enzimas y glicoproteínas de la cubierta viral, respectivamente) y varios genes codificantes para proteínas reguladoras (tat, rev, vif, vpu, nef y vpx).
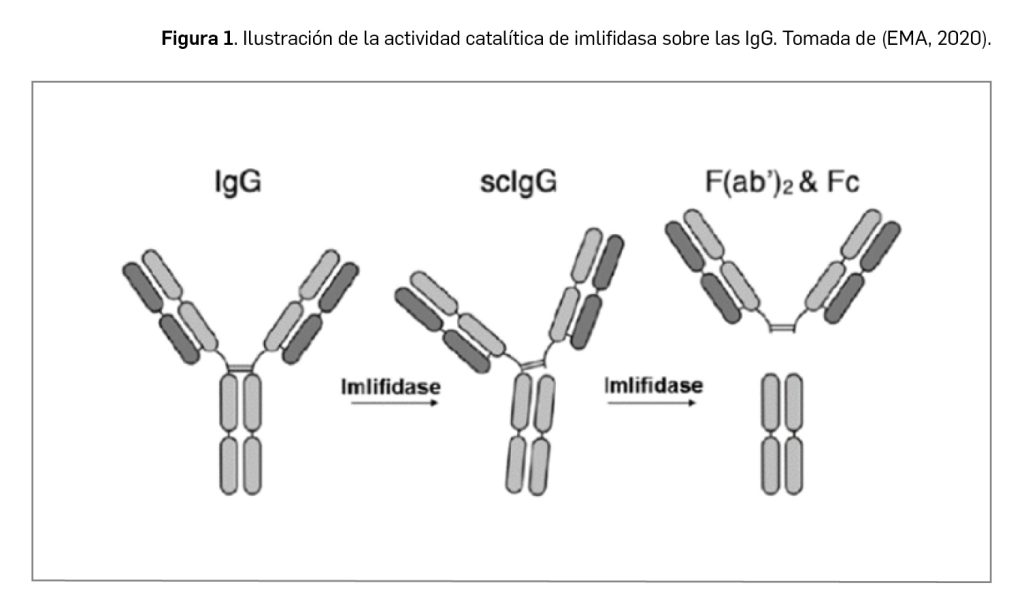
Los VIH son virus de geometría esférica (Figura 1). Su capa más externa –cubierta– está formada por una membrana lipídica derivada de la célula hospedadora, con un alto contenido en lipoproteínas, y en la que se insertan los antígenos de histocompatibilidad (HLA) del hospedador, que le permitirán una primera unión a la célula humana, y multitud de complejos heterodiméricos de glucoproteínas, incluidas gp120 y gp41. Por debajo de la membrana lipídica se encuentra la matriz proteica formada por la proteína p17, que recubre a la cápsula o cápside propiamente dicha, constituida por la proteína p24, y en cuyo interior se encuentran el material genético, la nucleoproteína y algunas enzimas (entre otras, la transcriptasa inversa y la integrasa). Con la participación de todos esos elementos, el virus se apodera de la maquinaria celular y si-lencia la replicación de numerosos genes celulares en favor de la replicación de los propios.
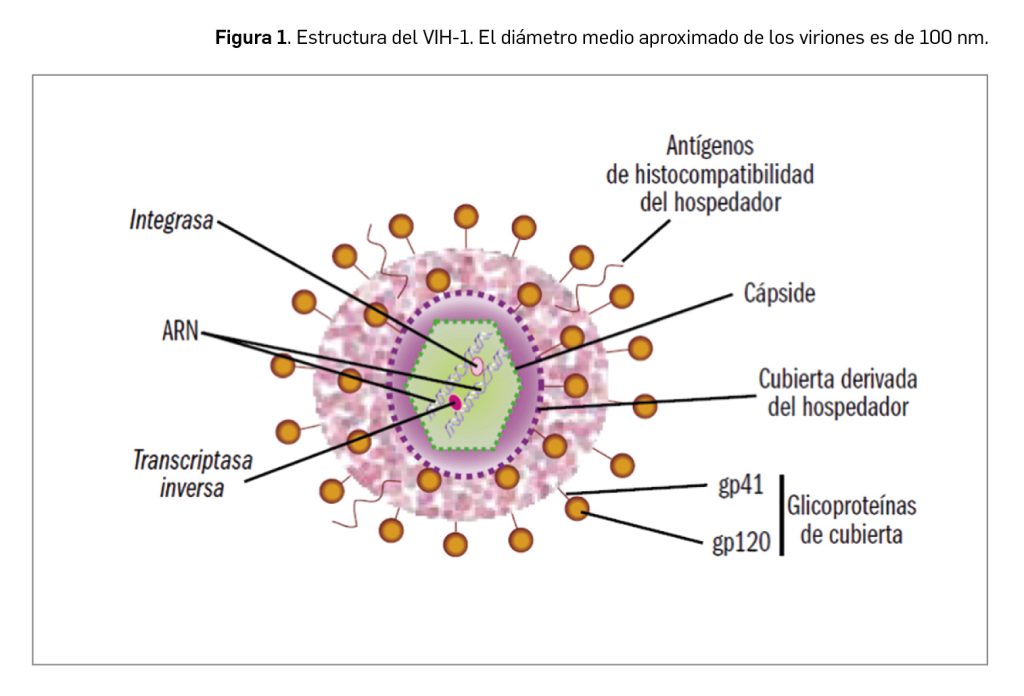
Se han descrito 2 tipos de VIH: VIH-1, el más prevalente en humanos, derivado del virus de la inmunodeficiencia de los simios de los chimpancés, y el VIH-2, que se originó a partir del retrovirus que infecta a los primates denominados mangabeys grises. Se localizan mayoritariamente en individuos de diversos países de África Occidental y otras áreas con lazos históricos, como la India. Dentro de cada uno de esos dos grandes grupos de VIH, se han descrito numerosos subtipos1, con características biológicas y patogenicidad distintas, algunos de los cuales han evolucionado mayoritariamente de forma pandémica.
En líneas generales, se trata de un virus extraordinariamente sensible a las condiciones ambientales, que no puede sobrevivir fuera del torrente sanguíneo o del tejido linfático, presentando especial tropismo por los macrófagos y los linfocitos T CD4+. Por ello, la transmisión del VIH entre personas se produce por vía sexual (80% de los contagios), parenteral y vertical (madre-hijo), mediante fluidos biológicos en los que el virus se mantiene viable, sobre todo sangre y secreciones (vaginal, espermática, etc.) que entren en íntimo contacto con estructuras potencialmente receptoras, como los vasos sanguíneos, o pequeñas erosiones en la piel o las mucosas.
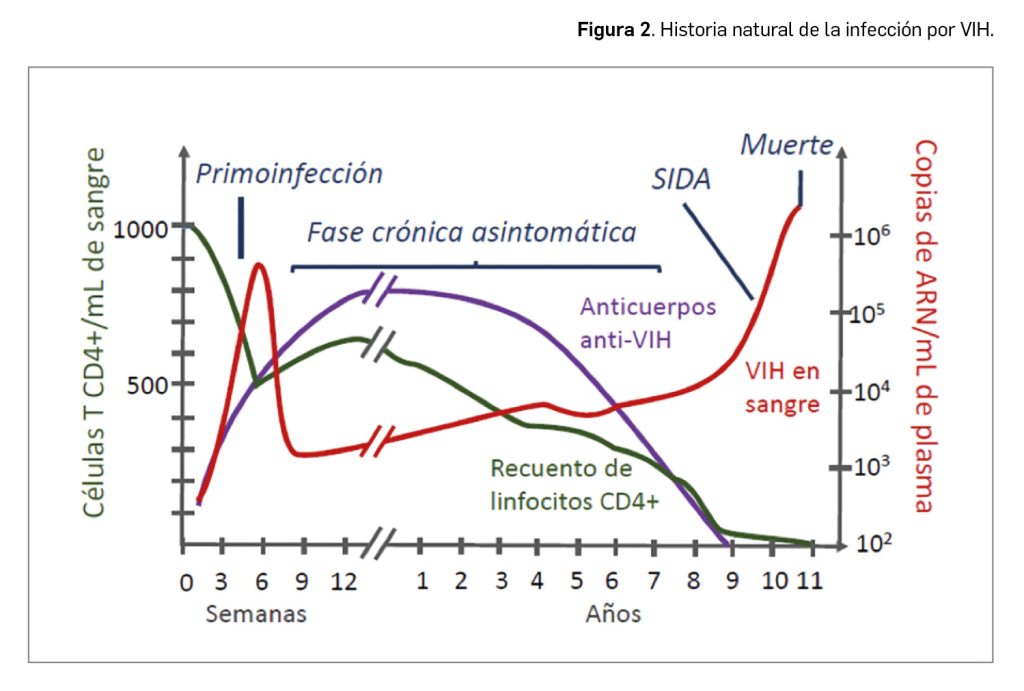
La respuesta inmunitaria frente al VIH se produce tanto en la vertiente humoral (intensa producción de anticuerpos contra las proteínas reguladoras y estructurales, así como de complemento e interferones) como en la celular (activación de poblaciones linfocitarias T colaboradoras, citotóxicas y de estirpe natural killer). Inicialmente, se produce una intensa expansión clonal de linfocitos CD8+ con actividad citotóxica dirigida frente a diversas proteínas del virus, que provoca cierta supresión de su replicación y una drástica disminución de la viremia, pudiendo incluso hacerse indetectable en sangre (< 50 copias de ARN/ml). Entre 3 y 5 semanas después de la infección aparecen los anticuerpos y a partir de ese momento el paciente se transforma en seropositivo. Aunque la respuesta llega a contener su replicación, es incapaz de erradicar el virus, que con el tiempo se difunde por todo el organismo.
Tanto el VIH-1 como el VIH-2 son capaces de evolucionar a la fase de infección crónica asintomática, a lo largo de la cual se produce un deterioro progresivo del tejido linfoide y, finalmente, si no se instaura tratamiento2, desemboca en la fase de SIDA (síndrome de inmunodeficiencia adquirida), que se define por un recuento de < 200 linfocitos T CD4+/ml de sangre o por la asociación de cualquier manifestación grave, como infecciones oportunistas (por ejemplo, por micobacterias, Pneumocystis jiroveci, Candida albicans, herpes simple y zóster, citomegalovirus, etc.) o ciertas patologías malignas (por ejemplo, ciertos tumores como sarcoma de Kaposi y ciertos linfomas).
Para un mayor detalle sobre la historia natural de la enfermedad (Figura 2) se recomienda consultar el artículo monográfico publicado en Panorama Actual del Medicamento (Fernández-Moriano, 2018).
Los datos publicados por la OMS en 2022 reflejan que a finales de 2021 había unos 38,4 millones de personas de todos los rangos de edad que vivían infectados con el VIH en todo el mundo (incluidos unos 2 millones de niños menores de 15 años), más de dos tercios –casi 26 millones– en el continente africano3, especialmente en África Subsahariana, región en que la tasa media de prevalencia del VIH se aproxima al 5% y alcanza el 25% en algunos países. Así, el VIH sigue siendo uno de los mayores problemas para la salud pública global: desde que se iniciara la epidemia en 1981 ha sido la causa de más de 40 millones de defunciones, de las cuales 650 000 se produjeron en 2021; el virus ha infectado en total a hasta 79 millones de personas, produciéndose en 2021 aproximadamente 1,5 millones de nuevos contagios. En todo caso, la epidemia se ha estabilizado o está mostrando signos de recesión en los últimos años: en dos décadas se ha reportado una significativa reducción de la incidencia desde los 3,4 millones de nuevas infecciones a nivel global en 1996 hasta los 1,8 millones en 2017, la mayoría de los cuales se concentran en las poblaciones de riesgo (mujeres en el continente africano, hombres homosexuales, prostitutas o drogadictos).
Pese a los problemas de acceso en determinados territorios4, los progresos en la prevención y en efectividad del tratamiento antirretroviral han permitido también reducir en más de la mitad el número de defunciones por enfermedades relacionadas con el SIDA a nivel mundial, desde el pico de 1,9 millones en 2004 hasta las 940 000 muertes en 2017.
Tratamiento antirretroviral (TAR)
Mientras no se disponga de fármacos erradicadores, el objetivo fundamental del TAR es suprimir la replicación del VIH para mantener la carga viral plasmática (CVP) en niveles indetectables durante el máximo tiempo posible. El estándar terapéutico es una CVP < 50 copias de ARN/ml de sangre, objetivo que persigue restaurar la función inmunitaria y limitar el desarrollo de resistencias virales para revertir la morbi-mortalidad asociada, mejorar la calidad de vida del paciente (para que su esperanza de vida se aproxime a la de la población general, lo cual actualmente se consigue eficazmente) y prevenir la transmisión del VIH.
El Documento de Consenso GeSIDA/Plan Nacional sobre el Sida respecto al tratamiento antirretroviral en adultos infectados por el virus de la inmunodeficiencia humana (Palacios et al., 2023) establece, en base a diversos estudios observacionales y clínicos, que el TAR debe iniciarse tan pronto como sea posible en todos los pacientes con infección por VIH-1 confirmada, con o sin sintomatología, independientemente del número de linfocitos T CD4+ y del valor de la CVP, si bien estos parámetros sí deben determinarse previamente; la excepción serían los pacientes que mantienen CVP indetectable de forma mantenida sin TAR (controladores de élite). El inicio precoz del TAR –idealmente en los primeros 14 días– se relaciona con una menor frecuencia de transmisión del VIH y de nuevas infecciones, y con menor incidencia de manifestaciones por infecciones o procesos tumorales. La farmacoterapia de esta infección ha progresado notablemente en las últimas décadas para mejorar la potencia, la tolerancia y la disponibilidad, asociándose con una disminución drástica en la morbimortalidad por SIDA, a lo que han contribuido las mejoras galénicas en las formulaciones de medicamentos que han permitido superar posologías complejas (varias tomas orales al día) y facilitar la adherencia a los tratamientos. A día de hoy, se dispone en España de numerosas opciones farmacológicas para el tratamiento de adultos con infección por VIH-1 (Fernández-Moriano, 2018). La elección preferente para el TAR de inicio en pacientes no tratados previamente se basa en combinaciones de, al menos, 2 o 3 fármacos (Tabla 1). Su uso combinado combate la supervivencia de cepas resistentes y, aunque se han estudiado combinaciones eficaces con menor número de fármacos, las autoridades sanitarias no las han autorizado aún como TAR de inicio. Las combinaciones de 3 fármacos indicadas constituyen también el tratamiento de inicio de elección de la infección crónica por VIH-1 (> 6 meses tras la primoinfección), fase en la que rige igualmente la recomendación de iniciar el TAR en todos los pacientes diagnosticados.
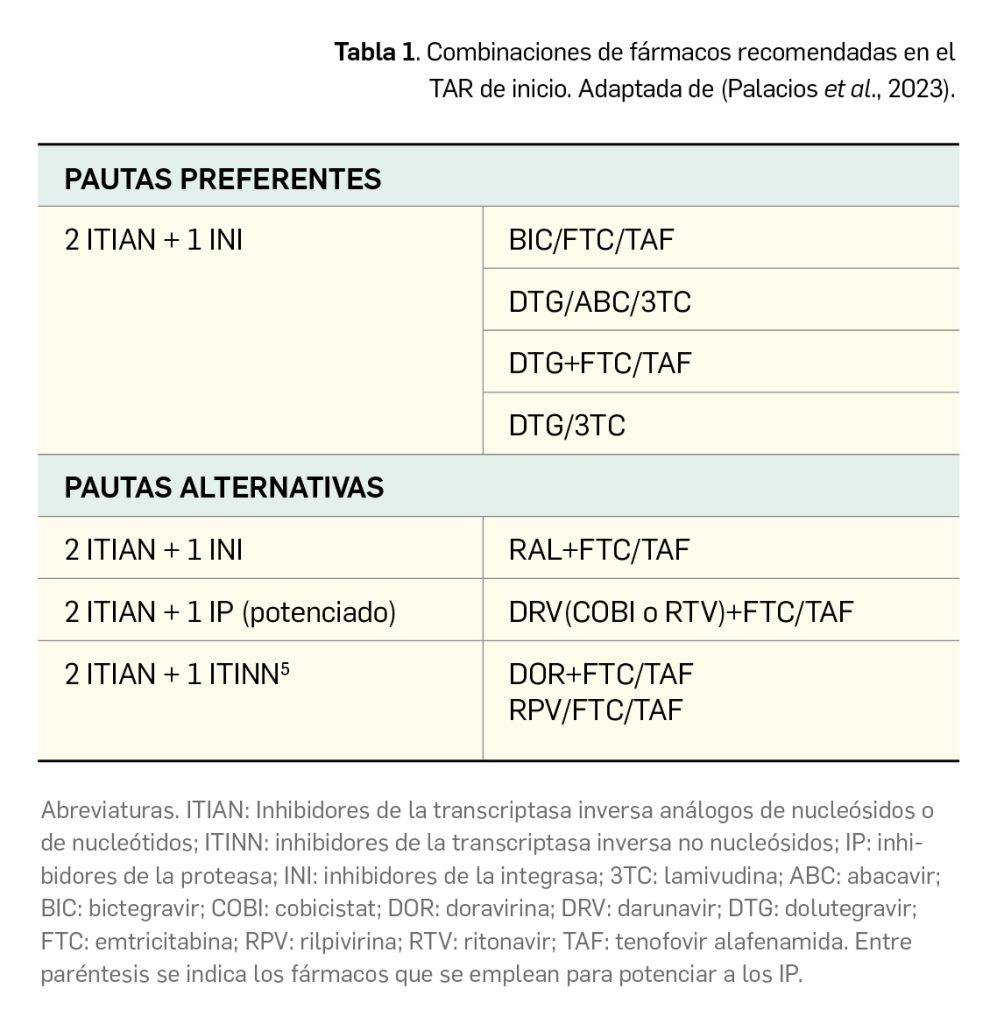
Para seleccionar una u otra familia de fármacos se valorarán las ventajas que aportan: el potencial de interacciones farmacológicas (menor en los INI, seguido de ITINN y mayor en los IP), la mayor barrera genética frente a resistencias (en los IP) y el menor coste (de los ITINN). Una pauta con dos ITIAN (preferentemente tenofovir/emtricitabina) y un INI (dolutegravir o bictegravir) puede tener la ventaja de una mayor concentración en las secreciones genitales para reducir más rápidamente la CVP durante las primeras 4‐8 semanas en comparación con los IP o ITINN, facilitando la reducción de los contagios por vía sexual. No se recomiendan como tratamientos de inicio pautas libres de ITIAN ni monoterapia con IP potenciados. Es fundamental recordar que, una vez iniciado, el TAR debe administrarse por tiempo indefinido, reevaluando periódicamente (dependiendo de comorbilidades, riesgo de interacciones y resistencias, mala adherencia, etc.) la pauta farmacológica administrada a partir de los 6 meses, cuando la infección se considerará crónica.
Diversas circunstancias pueden obligar a un cambio proactivo del TAR incluso en pacientes virológicamente suprimidos, tales como problemas de seguridad o tolerabilidad o la conveniencia de simplificar el régimen de tratamiento, pues suele asociarse con la toma frecuente de diversos comprimidos por vía oral; factores estos que pueden comprometer una adecuada adherencia terapéutica, fundamental para alcanzar los objetivos clínicos y minimizar la aparición de mutaciones de resistencia. En este sentido se han desarrollado varios enfoques para simplificar el TAR, entre los que sobresale la reducción del número de fármacos, habiéndose mostrado algunas combinaciones de 2 fármacos (por ejemplo, rilpivirina con dolutegravir, o dolutegravir con lamivudina) similares en eficacia a otros regímenes triples.
En cualquier caso, durante el tratamiento de por vida la aparición de resistencias –por acumulación de mutaciones virales– es un fenómeno inevitable, muchas veces consecuencia de terapias subóptimas, que limita la eficacia de nuevos regímenes. Su detección por métodos genotípicos es muy útil en casos de fracaso virológico (cuando la viremia sube por encima de 200 copias de ARN/ml), orientando la prescripción de terapias de rescate, si bien no se recomienda suspender el TAR en pacientes con fracaso virológico avanzado y sin opciones terapéuticas de rescate.
Son frecuentes los casos de pacientes infectados por virus que muestran resistencia cruzada a muchos fármacos de una misma familia, pues comparten el tipo de mutaciones que implican disminución o pérdida de la sensibilidad antiviral. En pacientes con antecedentes de fracaso, se recomienda incluir 2-3 fármacos activos en la nueva combinación, incluyendo en la medida de lo posible un fármaco de una nueva familia que permita evitar el fracaso virológico y las mutaciones de resistencia que surgirían en caso de monoterapia funcional.
Se considera infección multirresistente la de aquellos pacientes con virus que muestran resistencia fenotípica o genotípica6 bien a las tres familias principales de antirretrovirales (ITIAN, ITINN e IP) o bien a todas las familias estándar de fármacos (ITIAN, ITINN, IP, y/o INI). En la actualidad la proporción de pacientes con escasas opciones de tratamiento por multirresistencia del VIH se ha reducido notablemente, hasta menos del 2% (desde el 14% en 2007), pero estos siguen siendo vulnerables al fracaso terapéutico y a la progresión de la patología, con peor tolerancia a las limitadas opciones, y constituyen uno de los principales retos terapéuticos.
En ese contexto, la única opción que aumenta la probabilidad de supresión virológica suele ser la de introducir fármacos de familias nuevas, con mecanismos de acción novedosos y sin resistencia cruzada, que retengan actividad antiviral (medido por sensibilidad genotípica) y permitan construir un régimen de rescate que idealmente consiga, además de la supresión virológica, el aumento de los niveles de células CD4+ (que tiene también un impacto alto en la mortalidad y calidad de vida). El abordaje de estos pacientes siempre debe ser individualizado, teniendo en cuenta factores como los antecedentes de infecciones oportunistas y de uso de antirretrovirales, incluida sus toxicidades, el grado de inmunosupresión, el riesgo de progresión o las mutaciones de resistencia.
En los últimos años varios fármacos han demostrado eficacia –reducción del ARN viral– y tienen indicación aprobada en pacientes altamente pretratados y con resistencia a múltiples clases de antirretrovirales: dolutegravir, lenacapavir e ibalizumab7; estos dos últimos no están disponibles aún en España. No es posible hacer comparaciones directas entre ellos, y, en ocasiones, se hace necesario su uso en combinación.
Acción y mecanismo
Fostemsavir es un nuevo antirretroviral en forma de profármaco (sin actividad antiviral significativa) que se hidroliza en nuestro organismo por acción de la fosfatasa alcalina en la luz del tracto gastrointestinal a la fracción activa temsavir: este se une directamente a la subunidad gp120 de la glicoproteína gp160 de la envoltura del VIH-1 y, así, es capaz de inhibir selectivamente la interacción entre el virus y el receptor celular CD4, evitando la entrada del virus y la infección de células huésped CD4+. El medicamento ha sido autorizado para el tratamiento oral diario, en combinación con otros antirretrovirales, de adultos infectados por el VIH-1 multirresistente para quienes, de otro modo, no es posible establecer un tratamiento antirretroviral supresor.
Los estudios preclínicos permitieron demostrar que temsavir se une a la subunidad gp120 en un lugar cercano al sitio de unión al receptor CD4 y provoca que esa glicoproteína se mantenga en una conformación “cerrada”, lo que impide la interacción inicial entre el virus y los receptores celulares CD4, evitando cualquier paso adicional necesario para la entrada del virus en las células huésped. Los estudios con cultivos de células mononucleares de sangre periférica han demostrado que el fármaco tiene una actividad antiviral variable frente a los distintos subtipos del VIH-1, con valores de CI50 que oscilan entre 0,01 y >2000 nM (valor medio de 1,37 nM), pero tiene actividad reducida frente al subtipo AE del grupo M y carece de efecto frente a los grupos O y N del VIH-1 (porque contienen una alta frecuencia de polimorfismos concretos en el genoma) y frente al VIH-2. De manera interesante, no se ha observado actividad antagónica del nuevo fármaco con la de todos los antirretrovirales usados frente a la infección por VIH, y los fármacos sin actividad inherente contra el VIH (entecavir, ribavirina) no tienen capacidad de influir sobre el efecto de temsavir.
Aunque in vitro se han identificado ciertos fenotipos virales que tienen mutaciones en el gen codificante para gp120 con potencial relevancia clínica, temsavir continuó siendo activo contra los virus independientes de CD4 provenientes de laboratorio (AEMPS, 2023). Tampoco hubo evidencia de resistencia cruzada a fármacos representativos de otras clases de antirretrovirales, conservando temsavir actividad frente a virus resistentes a raltegravir; a rilpivirina o efavirenz; a abacavir, lamivudina, tenofovir o zidovudina; a atazanavir y darunavir; y a maraviroc o a enfuvirtida.
En estudios clínicos con pacientes muy pretratados se han podido identificar ciertos polimorfismos de gp120 (S375H, M426L, M434I o M475) que se asocian con una resistencia parcial del virus a temsavir, lo que se traducía en una menor reducción del ARN viral y menor proporción de sujetos con respuesta adecuada tras 8 días de tratamiento, aunque la evidencia disponible no permite extrapolar esos hallazgos a la eficacia antiviral en la población diana8.
Aspectos moleculares
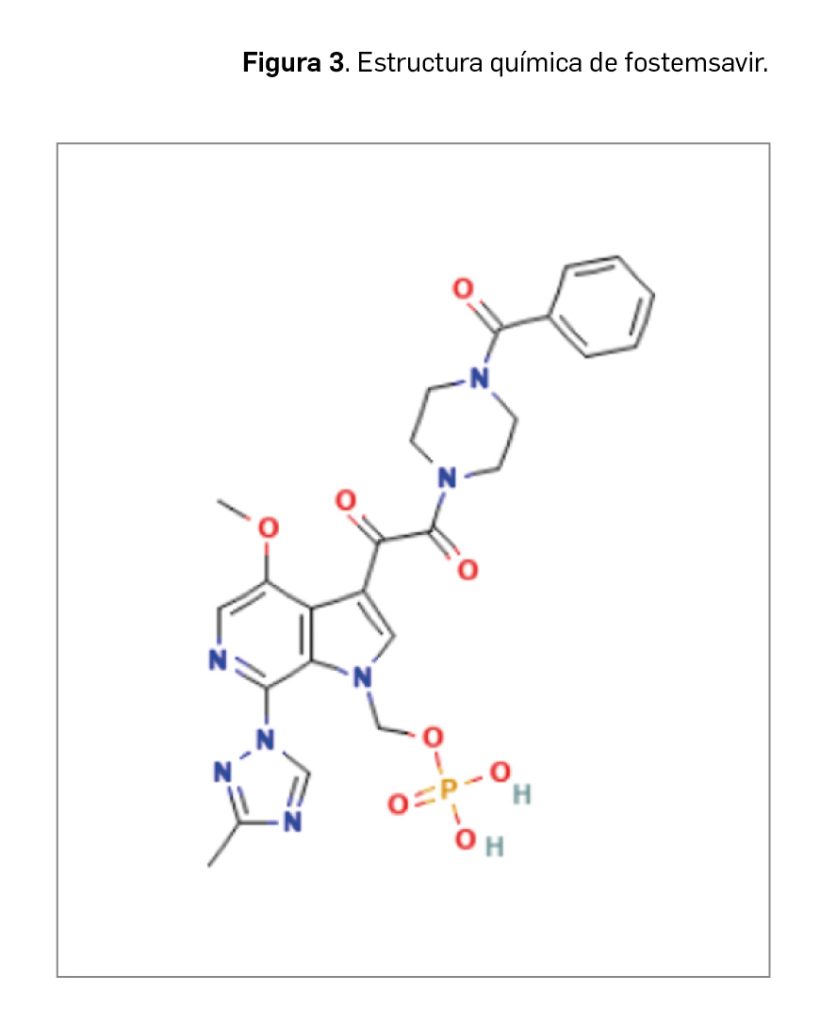
El nombre químico del nuevo fármaco en su forma de sal fostemsavir trometamina es (3-((4-benzoil-1-piperazinil)(oxo)acetil)-4-metoxi-7-(3-metil-1H-1,2,4-triazol-1-il)-1H-pirrolo[2,3-c]piridin-1-il)metil dihidrógeno fosfato, 2-amino-2-(hidroximetil)-1,3-propanodiol (1:1). Su fórmula C25H26N7O8P.C4H11NO3 se corresponde con un peso molecular relativo de 704,6 g/mol.
El principio activo se presenta como un polvo blanco o casi blanco ampliamente soluble en medios acuosos a pH de ≥ 3,7, pero menos soluble a pH más ácidos (en torno a 1,7) en que se convierte en el ácido libre. Su estructura (Figura 3) no contiene ningún centro quiral ni potencial de isomerismo geométrico. In vivo se transforma en temsavir tras la división de un grupo fosfono-oximetilo por acción de la fosfatasa alcalina en la superficie luminal del intestino delgado.
Eficacia y seguridad clínicas
La aprobación de fostemsavir en su indicación y pauta oral (600 mg dos veces al día) se ha basado fundamentalmente en la evidencia generada por un ensayo pivotal de fase 3 (estudio BRIGHTE), con diseño multicéntrico y multinacional (108 centros de 22 países), parcialmente aleatorizado, doblemente ciego y controlado por placebo, que incluyó a un total de 371 pacientes con infección por VIH-1, altamente pretratados y con fracaso virológico (≥ 400 copias ARN viral/ml de sangre) con la terapia vigente en el momento de la inclusión. Todos ellos debían carecer total o parcialmente de alternativas para alcanzar un tratamiento antirretroviral de éxito, por haber mostrado resistencia, intolerancia, contraindicación o rechazo al uso. Se excluyeron pacientes con patologías cardiacas o antecedentes de las mismas.
Según el número de fármacos activos que pudieran usarse en su régimen de TAR, el estudio constó de dos cohortes: una aleatorizada y otra no aleatorizada. La que proporcionó la evidencia de mayor calidad sobre la eficacia del nuevo fármaco fue la primera, en que los 272 sujetos incluidos, quienes podían tener 1 pero no más de 2 opciones de fármacos activos (de 1 o 2 familias farmacológicas), fueron asignados al azar (3:1) a recibir, con enmascaramiento y de forma adicional a su TAR basal, bien fostemsavir (n= 203) o bien un placebo equivalente (n= 69) durante 8 días; posteriormente todos los sujetos recibieron en régimen abierto fostemsavir junto a un tratamiento de base optimizado. En la cohorte no aleatorizada se enrolaron 99 pacientes que recibieron desde el primer día un tratamiento abierto con fostemsavir más una terapia de base optimizada, que podía además incluir otros fármacos en investigación.
Dado que todos los objetivos fueron evaluados en la cohorte aleatorizada (el análisis de la cohorte no aleatorizada fue solo descriptivo), se puede destacar que las características demográficas y clínicas basales estuvieron bien balanceadas entre los dos brazos de tratamiento. Considerados en conjunto, sobresalen las siguientes: edad mediana de 48 años (solo 4% mayores de 65 años), 74% varones, 68% de raza caucásica, mediana de 99,5 células CD4+/mm3 al inicio (72% con < 200 células/mm3), el 85% tenían antecedentes –diagnóstico– de SIDA y el 67% habían estado más de 15 años en tratamiento antirretroviral. El número de regímenes de TAR previos fue de ≥ 5 en el 83% de los pacientes, describiéndose un máximo de 1 fármaco9 completamente activo en la primera terapia optimizada de más de la mitad de ellos (52%), mientras que otra proporción importante (42%) tenía 2 fármacos activos disponibles.
El análisis de los datos tomó como fracaso virológico a todos los sujetos sin registro de carga viral en los puntos de corte temporal relevantes o a quienes cambiaron la terapia optimizada de base por pérdida de eficacia. Los resultados en la población por intención de tratar expuesta de la cohorte aleatorizada (Kozal et al., 2020) revelan que, en el día 8 de tratamiento, se vio una disminución media ajustada respecto al inicio del nivel plasmático de copias de ARN viral (medida en log10) –variable primaria de eficacia– que fue estadísticamente significativa a favor de fostemsavir: reducción de -0,79 log10 copias/ml (IC95% -0,89 a -0,70) frente a un descenso de -0,17 log10 (IC95% -0,33 a -0,01) con placebo, lo que suponía una diferencia neta de -0,63 log10 a favor del fármaco (p < 0,0001). Así, en el día 8, el 68% y el 50% de los pacientes tratados con fostemsavir tuvieron, respectivamente, una reducción de la carga viral mayor a -0,5 y -1 log10 copias de ARN/ml (variables secundarias de eficacia), frente a solo el 19% y el 12%, respectivamente, en el grupo placebo. En general, no se vieron diferencias sustanciales en cuanto a la reducción de la carga viral en pacientes con subtipo B y no B del VIH, y se observó una respuesta solo parcialmente reducida frente a los subtipos A1 y AE.
Con el uso a más largo plazo de la terapia con fostemsavir (más el tratamiento de base optimizado) en la cohorte aleatorizada se obtuvieron los resultados reflejados en la Tabla 2 (Lataillade et al., 2020). Nótese que, hasta la semana 96, 59 pacientes habían interrumpido el estudio, principalmente por falta de eficacia y problemas de adherencia.
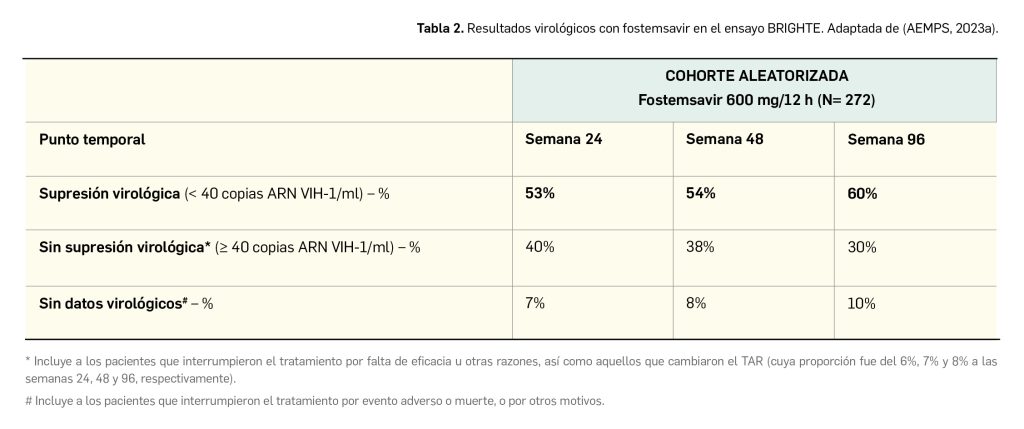
La carga viral inferior a 200 copias de ARN/ml se alcanzó en el 68%, 69% y 64% de los sujetos en las semanas 24, 48 y 96, respectivamente; los cambios medios en el recuento de células T CD4+ fueron aumentando con el tiempo, desde 90 células/mm3 en la semana 24 hasta 139 y 205 células/mm3 en las semanas 48 y 96, respectivamente. Los mejores resultados clínicos se alcanzaron con el uso conjunto de fostemsavir con dolutegravir y darunavir, mientras que el uso de fostemsavir sin dolutegravir se asoció con una menor eficacia.
Además, el análisis por subgrupos no mostró diferencias significativas en base a los diversos factores demográficos considerados (edad, raza, región geográfica, etc.), si bien la tasa de respuesta pareció estar asociada con las características basales de la enfermedad: la proporción de pacientes con supresión virológica durante el seguimiento fue menor en aquellos con una carga viral basal ≥ 100 000 copias ARN/ml (35-49% vs. 60-65% entre los que tenían < 100 000 copias/ml al inicio) o un recuento de CD4+ basal < 20 células/mm3 (32-46% vs. 58-61% entre los que tenían > 50 células/mm3).
En la cohorte no aleatorizada, que incluyó pacientes sin antirretrovirales totalmente activos y aprobados disponibles en el cribado (N= 99; mediana de edad de 50 años, 90% con diagnóstico de SIDA), la tasa de respuesta virológica fue del 37% en la semana 24, y se mantuvo en el mismo nivel hasta la semana 96, cuando se habían producido 38 interrupciones del estudio (sobre todo, por muertes). El incremento medio de células CD4+ en ese último punto temporal fue notablemente inferior que en la cohorte aleatorizada (119 vs. 205 células/mm3), con una proporción mayor de pacientes que cumplían los criterios de fracaso virológico (49% vs. 23% en la cohorte aleatorizada). Es preciso citar que 15 pacientes de esta cohorte fueron tratados con ibalizumab y tuvieron tasas de respuesta a la semana 24 mayores (53% vs. 35% entre los que no recibieron ese fármaco), si bien con el paso del tiempo la respuesta fue similar con independencia del uso de ese fármaco (33% a la semana 96 vs. 38% entre los que no lo usaron).
Adicionalmente, se tienen datos de un estudio de soporte, de fase 2b, aleatorizado y controlado, que incluyó a 254 pacientes infectados por VIH-1 y pretratados (aunque con menor exposición y resistencia a antirretrovirales que en el pivotal) y que comparó varias dosis orales de fostemsavir (400 mg/12 h, 800 mg/12 h, 600 mg/24 h y 1200 mg/24 h) frente a atazanavir+ritonavir, en ambos casos adicionados a un régimen de raltegravir y tenofovir. Tras 48 semanas, el estudio pasó a ser abierto y todos los pacientes de los brazos de fostemsavir pasaron a recibir la dosis más alta diaria.
Con datos de 251 pacientes tratados (unos 50 por brazo), los resultados reflejan una tasa de respuesta virológica (< 50 copias de ARN/ml) a la semana 24 del 69-80% con fostemsavir y del 75% en el grupo control. La eficacia a la semana 48 se mantenía en niveles similares y era comparable entre el tratamiento experimental y el control, también en lo referente al cambio medio frente al inicio en el recuento de células CD4+, y sin diferencias reseñables entre subgrupos. Aunque la proporción de abandonos era más baja con fostemsavir (32% vs. 41% con atazanavir), había mayor incidencia de abandonos por falta de eficacia (11% vs. 6% en el grupo control) (AEMPS, 2023b).
En cuanto a la caracterización de su perfil de seguridad, se tienen datos de casi 1500 individuos que recibieron al menos una dosis del nuevo fármaco durante su desarrollo clínico. En la cohorte aleatorizada del estudio pivotal, la mediana de exposición fue de 113 semanas, siendo los eventos adversos más comunes la diarrea, las náuseas, las infecciones del tracto respiratorio superior (incluida nasofaringitis y gripe), dolor de cabeza, pirexia, tos y bronquitis; en general fueron leves-moderados, limitados en el tiempo y se resolvieron sin necesidad de tratamiento. Los pacientes de la cohorte no aleatorizada registraron eventos adversos de mayor gravedad, así como discontinuaciones y muertes por eventos adversos.
Globalmente, las reacciones adversas al tratamiento más comunes fueron: diarrea (24%), cefalea (17%), náuseas (15%), erupción (12%), dolor abdominal (12%) y vómitos (11%). Entre los eventos considerados como graves (< 4%), durante el seguimiento a largo plazo en el estudio pivotal destacaron la neumonía, la celulitis y los eventos coronarios arteriales. Y entre los que supusieron discontinuación del tratamiento (7%, en su mayoría infecciones) se relacionaron con el tratamiento en el 3% de los pacientes, sobre todo alteraciones en el intervalo QT. Se registraron 36 muertes en el desarrollo de fostemsavir, una amplia mayoría debidas a complicaciones asociadas al SIDA y a infecciones agudas; solo una muerte por síndrome inflamatorio de reconstitución inmune se relacionó con el tratamiento, considerándose este como un evento adverso de especial interés, como también lo fueron las reacciones de hipersensibilidad, la prolongación del intervalo QT del electrocardiograma o el aumento de peso.
Aspectos innovadores
Fostemsavir es un nuevo antirretroviral en forma de profármaco que se hidroliza en nuestro organismo por acción de la fosfatasa alcalina en la luz del tracto gastrointestinal a la fracción activa temsavir. Este se une directamente a la subunidad gp120 de la glicoproteína gp160 de la envoltura del VIH-1 y provoca que esa glicoproteína se mantenga en una conformación “cerrada”: es así capaz de inhibir selectivamente la interacción entre el virus y el receptor celular CD4, evitando la entrada de los viriones y la infección de células huésped. En base a ello, y a la probada actividad antiviral de temsavir frente a distintos subtipos del VIH-1 (reducida frente al subtipo AE del grupo M y ausente frente a los grupos O y N), el medicamento ha sido autorizado para el tratamiento oral diario, en combinación con otros antirretrovirales, de adultos infectados por el VIH-1 multirresistente para quienes, de otro modo, no es posible establecer un tratamiento antirretroviral supresor.
Los datos clínicos que sustentan su aprobación de uso adicional a un régimen multifármaco de TAR oral derivan fundamentalmente de la cohorte aleatorizada de un estudio de fase 3 (N= 272 pacientes altamente pretratados y con limitadas opciones terapéuticas), considerada de adecuado diseño para valorar su eficacia a corto plazo (periodo controlado por placebo), pero que limita las conclusiones a largo plazo por no existir un brazo control más allá de los 8 primeros días de tratamiento. En dicho estudio se planteó la administración inicial del fármaco en combinación con el TAR en fracaso virológico, para después añadirlo al régimen optimizado con 1-2 agentes activos. En el día 8, el uso de una monoterapia funcional con fostemsavir demostró su eficacia significativamente superior a placebo (p < 0,0001), evidenciada en una reducción unas 4 veces mayor en la carga de ARN viral en sangre (-0,79 log10 vs. -0,17 log10 con placebo; descenso neto a favor del fármaco de -0,63 log10). No se vieron diferencias sustanciales entre subgrupos de pacientes ni tampoco en la eficacia antiviral en pacientes con subtipo B (dominante en España) y no B del VIH-1.
A más largo plazo, aunque no se puede comparar con un control, es destacable que el uso de fostemsavir con un régimen optimizado con 1-2 fármacos activos adicionales se asocia con una eficacia mantenida: la tasa de supresión virológica a la semana 96 es del 60%, aumentando desde el 53% a la semana 24. Se puede asumir que el uso concomitante de agentes con actividad total o parcial contribuyó a esos niveles de eficacia, que no pueden atribuirse exclusivamente a fostemsavir. De hecho, la coadministración mayoritaria de dolutegravir (84% de pacientes) se asoció con los mejores resultados clínicos; por contra, fostemsavir sin dolutegravir parece relacionarse con menor eficacia. De modo interesante, en la cohorte no aleatorizada, que incluyó pacientes sin antirretrovirales activos disponibles en el cribado (N= 99), la tasa de supresión virológica fue del 37% en la semana 96, aunque esa cifra también puede verse sobreestimada por el efecto de ibalizumab (no aprobado en España) en hasta 15 pacientes. La falta de datos comparativos a largo plazo, la gravedad de la enfermedad de base y el uso simultáneo de otros fármacos también dificultan las conclusiones sobre el perfil toxicológico de fostemsavir, y hacen necesarios estudios de farmacovigilancia para caracterizarlo. En principio, la seguridad del nuevo fármaco parece aceptable y manejable clínicamente, pues las reacciones adversas más comunes (> 10%) durante su uso se refieren a la tolerabilidad gastrointestinal (diarrea, náuseas, dolor abdominal y vómitos), reacciones de hipersensibilidad (erupción cutánea) y cefalea; las frecuentes infecciones del tracto respiratorio se asocian en mayor medida a la patología de base. Una amplia mayoría de los eventos adversos fueron leves-moderados en severidad y autolimitados durante el tratamiento, destacando entre las consideradas graves (< 4%) la neumonía, la celulitis y los eventos coronarios arteriales. La tasa de interrupciones (7%, solo 3% en relación con el tratamiento) se asocia esencialmente a infecciones, y solo una de las muertes durante los estudios se vinculó con el uso de fostemsavir, por un caso de síndrome inflamatorio de reconstitución inmune, un evento adverso de especial interés.
No se dispone de comparaciones directas con el resto de alternativas disponibles en España –lenacapavir y dolutegravir– para pacientes con infección por VIH-1, fracaso virológico y escasas/nulas opciones de tratamiento. Habida cuenta de sus posibles combinaciones en el TAR optimizado, las comparaciones indirectas son especialmente poco robustas en términos estadísticos, sobre todo por el distinto perfil de pacientes estudiados en los ensayos clínicos.
El IPT (AEMPS, 2023b) alude a una comparación indirecta ajustada de la población incluida en el estudio pivotal para evaluar el beneficio de fostemsavir junto a una terapia de base optimizada frente a pautas optimizadas distintas que no incluyesen dicho fármaco en otros estudios. Sus resultados apuntan a que la eficacia de un régimen con fostemsavir es similar a otro con ibalizumab en la probabilidad de alcanzar la supresión virológica o en el incremento de los niveles de linfocitos CD4+, y no parece aportar resultados superiores a otros TAR optimizados que combinen principalmente dolutegravir, darunavir y tenofovir. Otro reciente análisis indirecto sugirió que la adición de lenacapavir a una terapia de base optimizada se asocia con una probabilidad 6,6 veces mayor de supresión viral que la adición de fostemsavir, 8,9 veces mayor que la de ibalizumab y hasta casi 13 veces mayor respecto al uso de la terapia optimizada sola, si bien no hubo diferencias reseñables en el recuento de células CD4+ (Chatzidaki et al., 2023).
Por último, se puede aludir a los resultados del estudio VIKING-4 (Akil et al., 2014) que enroló a pacientes (n= 30) con VIH-1 multirresistente con fracaso a casi todas las familias de antirretrovirales disponibles (incluso con resistencia a otros inhibidores de integrasa), y probó que una pauta con dolutegravir (50 mg/12 h) provoca un descenso notablemente mayor que placebo al día 8 (diferencia neta de -1,16 log10 copias/ml), incluso en pacientes con resistencia a otros inhibidores de integrasa. A la semana 48, hasta el 40% de los pacientes tuvo carga viral indetectable, y no se vieron discontinuaciones por eventos adversos.
A pesar de las dudas sobre el riesgo de aparición de resistencias durante el tratamiento con fostemsavir (los datos apuntan a que podría tener una barrera genética menor que otros fármacos, lo cual podría afectar a la eficacia de un TAR optimizado) y de la necesidad de contar con resultados de eficacia y seguridad en condiciones de vida real, este inaugura una nueva clase de antirretrovirales que puede ser útil como régimen de rescate en pacientes infectados por cepas de VIH-1 multirresistentes. Aunque comparaciones indirectas apuntan a una mayor eficacia de lenacapavir, se debe tener en cuenta que ambos pueden formar parte del mismo régimen, dado que no se han encontrado por ahora resistencias cruzadas con otros fármacos antirretrovirales.
Fostemsavir es, por tanto, una opción más dentro del reducido arsenal farmacológico disponible para el tratamiento de las infecciones multirresistentes por VIH-1.