Resumen
Oritavancina es un nuevo lipoglicopéptido que ejerce su efecto antibacteriano por un triple mecanismo sobre la síntesis y reparación de la pared bacteriana (inhibe la transglicosilación y la transpeptidación e interrumpe la integridad de la membrana bacteriana), provocando un aumento de la permeabilidad en dicha pared que permite el contacto directo entre citoplasma bacteriano y el entorno químico externo. Ello resulta en una muerte rápida de las bacterias, que es la base de la autorización del medicamento en el tratamiento, en dosis única intravenosa y según las directrices oficiales del uso de antibacterianos, de pacientes adultos con infecciones agudas de la piel y tejidos blandos de la piel (IPTBs). Presenta un espectro de actividad –limitado– similar a otros glicopéptidos aprobados con igual indicación: microbicida frente a las bacterias grampositivas más frecuentemente implicadas en IPTBs, como Staphylococcus aureus (incluidas las cepas resistentes a meticilina, SARM) y los estreptococos betahemolíticos (como Streptococcus pyogenes) o del grupo viridans.
Su aprobación se sustentó en dos amplios ensayos de fase 3, de adecuado diseño, que probaron la no inferioridad de oritavancina frente a vancomicina (fármaco de elección muy usado frente a SARM) en términos de la tasa de curación clínica tras 7-14 días desde el fin del tratamiento: 80-83% con oritavancina y 80-81% con vancomicina; en pacientes tratados durante más de una semana, se alcanzó una tasa del 91-93% y 89-95%, respectivamente. No se puede concluir respecto a su eficacia frente a infecciones distintas a celulitis, abscesos e infecciones de heridas, en pacientes con septicemia, ni frente a estafilococos con sensibilidad reducida a vancomicina. Tampoco se han realizado comparaciones directas con otros antibióticos, pero los estudios comparativos indirectos disponibles –de robustez limitada– sugieren que oritavancina y dalvabancina, con un mejor balance beneficio-riesgo que telavancina, pueden considerarse como alternativas interesantes y similares a otros antibióticos usados en terapia empírica estándar cuando hay sospecha de SARM, incluido vancomicina.
Se trata de un fármaco relativamente bien tolerado, con un perfil de seguridad comparable al de otros glicopéptidos. La mayoría de las reacciones adversas al fármaco, más frecuentes en mujeres y en pacientes de > 75 años de edad, son leves-moderadas en severidad y no suponen interrupciones del tratamiento (< 4%). Por su frecuencia, destacan las reacciones de hipersensibilidad (12%, como prurito y urticaria), las náuseas (10%), el dolor de cabeza (7%), los vómitos (5%) y los eventos adversos hepáticos (5%); la celulitis es la reacción adversa grave más reportada (1%).
En resumen, oritavancina representará una alternativa más a otras opciones de antibioterapia ambulatoria (linezolid, tedizolid y dalvabancina) y por vía parenteral (vancomicina, linezolid, tedizolid, daptomicina) previamente disponibles. Su elevada vida media (200-300 h) puede ser una ventaja de cara a la adherencia terapéutica (dosis única), por ejemplo frente a vancomicina o teicoplanina, pero a la vez ser un inconveniente ante problemas de seguridad o cuando se busca el desescalado. Por tanto, en el tratamiento empírico de IPTBs parece más conveniente usar otras alternativas que puedan retirarse fácilmente cuando se conozca el agente causal. A pesar de no suponer ninguna mejora clínica sustancial en su indicación, la incorporación de nuevos agentes antibacterianos con actividad frente a microbios multirresistentes es siempre una buena noticia para la sociedad.
Aspectos fisiopatológicos
Las infecciones de piel y tejidos blandos de la piel (en adelante, IPTBs)1 son, en conjunto, uno de los tipos de infección más comunes en nuestro medio, constituyendo la tercera causa más frecuente de infecciones, tras las urinarias y las respiratorias, tanto en el ámbito comunitario como hospitalario. El último informe EPINE, divulgado en 2018, pone de manifiesto que este tipo de infecciones representan el 4,2% y el 10,6% del total de infecciones relacionadas con la asistencia sanitaria y comunitarias, respectivamente, excluyéndose de esa cifra las infecciones del lugar quirúrgico (SEMPSPH, 2018).
Las IPTBs son aún más frecuentes en las personas mayores que en la población general y, particularmente, en las personas hospitalizadas, debido a la coexistencia de varios factores de riesgo, tales como inmunodepresión, malnutrición, diabetes, otras afecciones crónicas e incontinencia fecal. Asimismo, en las residencias, en torno al 10% de los pacientes que siguen un tratamiento antibiótico lo realiza por una infección de esta tipología.
Entre los motivos por los que las personas mayores son más susceptibles a las infecciones de la piel y tejidos blandos cutáneos, destaca el hecho de que, en la edad avanzada, todas las capas de la piel se encuentran alteradas, debido fundamentalmente a una disminución en la velocidad de renovación de células y a la acumulación consiguiente de células muertas. Asimismo, el estrato córneo es más delgado, sobre todo en zonas expuestas, tiene menor contenido en agua y mayor velocidad de descamación, mientras que la epidermis también es más delgada y disminuye el número de glándulas sudoríparas en la dermis y la hipodermis.
Las infecciones de la piel más frecuentes, sobre todo en las personas mayores, son las de etiología bacteriana por grampositivos, el intertrigo, el herpes y la onicomicosis. El punto de entrada más frecuente de las IPTBs son las soluciones de continuidad por pequeñas heridas, aunque también lo pueden ser traumatismos o heridas quirúrgicas. La fiebre, el signo cardinal de las infecciones, está ausente o se presenta más tarde en el 20-30% de los casos, lo que retrasa el diagnóstico y aumenta la mortalidad de este tipo de enfermedades.
La etiología predominante varía según el tipo de infección y la localización, pero las bacterias que más frecuentemente ocasionan infecciones en la piel y tejidos blandos del anciano son las grampositivas, como estreptococos betahemolíticos del grupo A (Streptococcus pyogenes) y Staphylococcus aureus; las infecciones por gramnegativos y anaerobios son más raras. Si bien el estreptococo es más frecuente en la piel del anciano, en los pliegues cutáneos con humedad y maceración son más frecuentes las enterobacterias.
La presentación clínica de las IPTBs puede ser muy variada y compleja, desde cuadros leves como infecciones superficiales de la piel, pasando por infecciones moderadas como la celulitis o abscesos cutáneos, hasta procesos graves con afectación sistémica como la fascitis necrotizante o la gangrena de Fournier, que pueden requerir antibioterapia sistémica, hospitalizaciones y cirugía. En su clasificación suele simplificarse entre purulentas o no purulentas y, según su gravedad, en leves, moderadas o graves. Las infecciones bacterianas más frecuentes se describen a continuación.
- La erisipela es la infección bacteriana de la dermis, mientras que la celulitis también afecta al tejido celular subcutáneo y es menos demarcada. Se localiza mayoritariamente en las extremidades inferiores y, en segundo lugar, en la cara. Se manifiesta con eritema, tumefacción, calor y dolor. En la erisipela se forman con frecuencia ampollas superficiales, que pueden hacerse hemorrágicas o necróticas. Asimismo, una celulitis grave puede producir necrosis dérmica afectando a fascia y músculo subyacente. Los gérmenes causantes más frecuentes de la erisipela son los estreptococos betahemolíticos (A: Streptococcus pyogenes; B: Streptococcus agalactiae), aunque también pueden producirse por otros estreptococos o estafilococos. En la celulitis, la etiología suele ser más variada, con predominio de estreptococos, si bien pueden estar implicados neumococos y gramnegativos. Las complicaciones en ancianos son muy frecuentes cuando no se instaura tratamiento antibiótico: erisipela y celulitis pueden evolucionar a fascitis, necrosis y sepsis, y en las extremidades pueden producir linfedema, elefantiasis y trombosis venosa profunda. La celulitis orbitaria es también frecuente en ancianos y suele producirse por Streptococcus viridans (un estreptococo alfahemolítico), solo o asociado a gramnegativos.
- La fascitis necrosante es una infección particularmente destructiva que causa rápidamente la necrosis de tejidos profundos. La etiología más común es polimicrobiana e incluye cocos grampositivos y bacilos gramnegativos, y anaerobios, como Escherichia coli, Klebsiella pneumoniae y Pseudomonas aeruginosa. Alrededor del 10% está producida por Streptococcus pyogenes, y puede evolucionar a gangrena estreptocócica. Las lesiones menores, como los forúnculos, se encuentran en alrededor del 20% de los pacientes antes de que presenten una fascitis necrosante.
- El impétigo es una infección superficial de la piel, que se presenta sobre áreas de piel normal o eritematosa sobre la que aparecen lesiones vesiculoampollosas, de consistencia flácida y ligeramente pruriginosas. Se manifiesta como placas rojas, blandas, dolorosas, calientes y bien delimitadas, si bien en ancianos se presenta más comúnmente en forma ampollosa, producida por Staphylococcus aureus y Streptococcus pyogenes. El contenido de las ampollas es claro, seroso o purulento. En el impétigo ampolloso el estado general suele ser bueno y sin fiebre; son raras las complicaciones como la linfadenitis o la glomerulonefritis.
- El ectima es una infección vesiculopustulosa, que posteriormente se abre y forma una úlcera de márgenes elevados indurados y violáceos, más profunda que en el impétigo. Se localiza casi siempre en las extremidades inferiores, donde las lesiones ampollosas dejan a la luz unas úlceras de fondo granuloso, exudativo, purulento y con bordes indurados. Evoluciona favorablemente a pesar de la presencia de linfangitis, fiebre, adenopatías y malestar general.
- Las foliculitis son infecciones superficiales o profundas que se asientan alrededor de un folículo piloso. Se localizan en cualquier zona de pelo corto y grueso, como cuero cabelludo, barba, axilas, nalgas y extremidades inferiores. Son lesiones pustulosas de pequeño tamaño, alrededor de un pelo y rodeadas de un halo eritematoso y ligeramente pruriginosas. Su etiología más frecuente es Staphylococcus aureus, pero pueden estar involucrados estreptococos y gramnegativos. Por su parte, el furúnculo es una foliculitis aguda, profunda y necrosante, que aparece en zonas pilosas de roce o presión, se inicia como un nódulo duro, rojo y doloroso, crece rápidamente de tamaño y se rodea de un edema considerable y es muy doloroso; puede acompañarse de fiebre y malestar general, e incluso ocasionar complicaciones locales (linfadenitis, linfangitis) o generales (osteomielitis, endocarditis, sepsis). Finalmente, el ántrax consiste en una foliculitis profunda que se produce sobre todo en pacientes diabéticos, malnutridos y con malas condiciones de higiene, en quienes suele afectar a la raíz de varios pelos (en especial, los de la nuca y/o espalda), así como al tejido dérmico y subcutáneo de alrededor, con la formación de varios focos de necrosis; se presenta como una placa elevada, edematosa, roja, caliente y dolorosa, que se acompaña de fiebre elevada, dolor intenso y malestar general. Los factores que favorecen la aparición de estas lesiones son: una alta carga nasal de colonizados por Staphylococcus aureus resistentes a meticilina (SARM), arañazos, diabetes mellitus, obesidad, neoplasias linfoproliferativas, desnutrición o tratamiento previo con glucocorticoides o inmunosupresores.
- Por último, el eritrasma es una infección crónica que se da más en mayores hospitalizados. Está causada por Corynebacterium minutissimum y se presenta como una placa delimitada de forma variable, que al inicio es húmeda y roja, y tiende a volverse marrón y descamativa. Se localiza en pliegues interdigitales de pies, ingles o axilas.
El riesgo de empeoramiento y la mortalidad asociada a muchas de estas infecciones agudas requieren la administración rápida –empírica– de terapia antibacteriana sistémica, y en la mayoría de los casos manejo quirúrgico y hospitalización para minimizar el daño tisular y evitar la diseminación de la infección. No obstante, en un estudio con más de 47.000 pacientes hospitalizados por IPTBs se observó que el fallo del tratamiento ocurre en casi un cuarto de los pacientes (22,8%) y se asocia con un aumento de la mortalidad en casi 3 veces (OR= 2,91); se constató que esa alta tasa de fracaso terapéutico también prolonga la duración tanto de la terapia intravenosa como de la estancia en el hospital (de 5 a 6 días) (Edelsberg et al., 2008).
Otra problemática importante en el abordaje de estas infecciones se refiere al aumento notable durante las últimas décadas en la incidencia de IPTBs por grampositivos multirresistentes como el Staphylococcus aureus resistente a meticilina (SARM) y el neumococo (Streptococcus pneumoniae) resistente, lo cual está obligando a cambiar el perfil clásico del tratamiento antibiótico, pues frente a dichos microbios se inhabilita el uso de la mayoría de los antibióticos considerados de elección en este tipo de infecciones (sobre todo, los betalactámicos).
El caso del SARM es especialmente preocupante, pues sus cepas se han difundido a escala mundial. Se han descrito numerosos mecanismos de resistencia en S. aureus, incluyendo la trasmisión de la misma a través de plásmidos compartidos entre bacterias o la transferencia desde distintos genes de la bacteria. Su prevalencia varía notablemente según zona geográfica, incluso dentro del mismo país. En Europa se han descrito cifras de prevalencia de este patógeno en IPTBs variables entre el 10,2% y el 22,8%, llegándose a describir un 25,3% de aislamientos de SARM invasivo en España en el año 2017; en EE.UU. esa prevalencia es mayor, pudiendo alcanzar la mitad de los casos de IPTBs. Debe tenerse en cuenta que SARM no solo tiene relevancia clínica en el ámbito hospitalario, sino también en las infecciones adquiridas en la comunidad: por ejemplo, un estudio observacional prospectivo realizado en un Servicio de Urgencias de la Comunidad de Madrid en 2010 reveló ya entonces que su prevalencia en pacientes que ingresaron con una infección cutánea supurativa adquirida en la comunidad a lo largo de un semestre era del 22%, y entre el total de infecciones estafilocócicas, representaba el 33% (Casado et al., 2010).
No existe un consenso generalizado sobre el tratamiento antibiótico empírico a implementar frente a las IPTBs, si bien el uso de antibióticos de la familia de los betalactámicos se reconoce todavía como una de las mejores opciones en los casos en los que no se sospecha de que esté implicado SARM. Las recomendaciones terapéuticas más aceptadas a nivel internacional están basadas en la presencia o no de exudado purulento y en la gravedad de la infección, recomendándose solo en las infecciones purulentas severas el uso empírico de antibióticos que retienen su actividad frente a SARM, tales como vancomicina, daptomicina, linezolid, ceftarolina, telavancina2 o el de más reciente aprobación dalbavancina.
Junto a la resistencia a meticilina, un porcentaje creciente de cepas de SARM están adquiriendo la condición de multirresistentes, volviéndose insensibles a múltiples fármacos antibacterianos, incluyendo a algunos de los más recientemente incorporados, como oxazolidinas (linezolid, tedizolid), estreptograminas, pleuromutilinas (tiamulina), etc. Además, las terapias disponibles actualmente tienen ciertas limitaciones: todas ellas consisten en regímenes multidosis y de varios días de duración, lo que implica que los pacientes estén hospitalizados durante el tratamiento y expuestos a un mayor riesgo de adquirir y difundir infecciones nosocomiales; algunas requieren ajustes posológicos en pacientes con insuficiencia renal y monitorización terapéutica (vancomicina); con otras se deben extremar las precauciones al usarlas en mujeres embarazadas (tigeciclina); y otras incluso pueden provocar reacciones adversas relacionadas con la inmunosupresión (linezolid).
Todo ello ha hecho que el SARM sea considerado como una prioridad entre los microorganismos importantes para la investigación y desarrollo de nuevos antibióticos, y que se hayan impulsado diferentes iniciativas encaminadas a la concienciación del problema sanitario que representa, en especial en el Sur y Este de Europa, donde la prevalencia de cepas de SARM es mayor. En este sentido, la adecuada adherencia terapéutica a los regímenes prescritos es fundamental para limitar el potencial desarrollo de resistencias microbianas.
Por otra parte, los estreptococos y, en especial Streptococcus pyogenes (estreptococo beta-hemolítico del grupo A), son microorganismos habitualmente implicados en las IPTBs. No obstante, al contrario que en el caso de S. aureus, su relevancia clínica y terapéutica radica más en su virulencia que en su resistencia a antimicrobianos, pues hoy en día sus cepas son uniformemente sensibles a penicilina y otros betalactámicos.
Acción y mecanismo
Oritavancina es un nuevo antibiótico semisintético perteneciente al grupo de los lipoglicopéptidos que ejerce su efecto antibacteriano a través de un mecanismo de acción complejo y triple: inhibe el paso de polimerización –transglicosilación– de la biosíntesis de la pared de la célula bacteriana mediante su unión al péptido madre precursor de los peptidoglicanos, inhibe la etapa de enlazamiento cruzado –transpeptidación– de la biosíntesis de la pared celular por su unión a los segmentos de formación de puentes peptídicos y, en su forma dimerizada, provoca la disrupción de la integridad de la membrana bacteriana conduciendo a la despolarización y permeabilidad incrementada, todo lo cual resulta en una muerte rápida de la célula. En base a ello, el medicamento ha sido autorizado para su uso a nivel hospitalario por vía intravenosa, siguiendo las directrices oficiales del uso apropiado de antibacterianos, en el tratamiento de pacientes adultos con infecciones bacterianas agudas de la piel y tejidos blandos de la piel.
Es preciso recordar que la cubierta de las bacterias varía sustancialmente de una especie a otra, pero colectivamente se aprecian diferencias cualitativas entre bacterias grampositivas y gramnegativas. Estas últimas disponen de una membrana citoplásmica similar a la de las células eucariotas: rodeando a la membrana celular existe lo que se conoce como espacio periplásmico, que a su vez está encerrado por una red de peptidoglucano y, finalmente, por una membrana exterior. En cambio, la cubierta de las grampositivas es más simple, pero contiene una capa mucho más tupida de peptidoglucano, formada por fibras poliméricas3 estrechamente entrelazadas que forman una estructura reticular cuya misión esencial es proporcionar rigidez a la cubierta bacteriana, y permite a la bacteria sobrevivir en entornos químicos agresivos. Dado que la osmolaridad citoplasmática bacteriana es de casi 3 veces la de las células eucariotas, sin la existencia de una pared celular y su red de peptidoglucano, el protoplasto bacteriano se hincharía y estallaría, debido a su hipertonicidad con respecto al medio (generalmente hipotónico), conduciendo a la muerte de la célula bacteriana. Se comprende, por tanto, que el efecto inhibitorio de oritavancina sobre la síntesis correcta de la pared bacteriana subyace tras su efecto bactericida.
En ensayos in vitro, el fármaco ha demostrado tener actividad microbicida4 frente a las bacterias grampositivas más frecuentemente implicadas en las IPTBs, o sea, S. aureus sensible a meticilina y resistente a meticilina (SARM), estreptococos beta-hemolíticos (de los grupos A, B, C y G) y estreptococos del grupo viridans (grupo S. anginosus). De igual modo, ha probado su actividad in vitro frente a enterococos, tanto sensibles como resistentes a vancomicina, si bien no se dispone de evidencia clínica que respalde su uso frente a cepas de enterococos o S. aureus resistentes a vancomicina, o con sensibilidad disminuida, frente a las que sí se vieron in vitro ciertos casos de resistencia. En los estudios clínicos se ha confirmado la eficacia antibiótica de oritavancina frente a los siguientes patógenos que mostraron sensibilidad in vitro: Staphylococcus aureus, Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, estreptococos del grupo anginosus (incluye S. anginosus, S. intermedius y S. constellatus).
Por el contrario, oritavancina no posee actividad frente a bacterias gramnegativas, y solo retiene actividad reducida frente a grampositivas que pertenecen a los géneros Lactobacillus, Leuconostoc y Pediococcus, todos los cuales son intrínsecamente resistentes a los glicopéptidos. No se conoce ningún caso de resistencia cruzada entre oritavancina y otras clases de antibióticos no glicopéptidos.
Aspectos moleculares
Oritavancina forma parte, junto a vancomicina (cabeza de serie), del grupo de antibióticos glicopeptídicos, todos los cuales comparten la presencia de un núcleo estructural heptapeptídico: un resto azucarado (aminoazúcar) unido a un sistema de tres anillos aromáticos ligados entre sí por puentes oxi (–O–) y con átomos de cloro (Cl); también hay una cadena peptídica simple y un sistema de bis-resorcinol. Más concretamente, oritavancina (Figura 1) tiene, como los ya comercializados teicoplanina y dalvabancina, una estructura más compleja que vancomicina por contener, además de aminoazúcares distintos, restos de ácidos grasos de diversa longitud (9 a 12 átomos de carbono) que le confieren ese carácter de lipoglicopéptido.
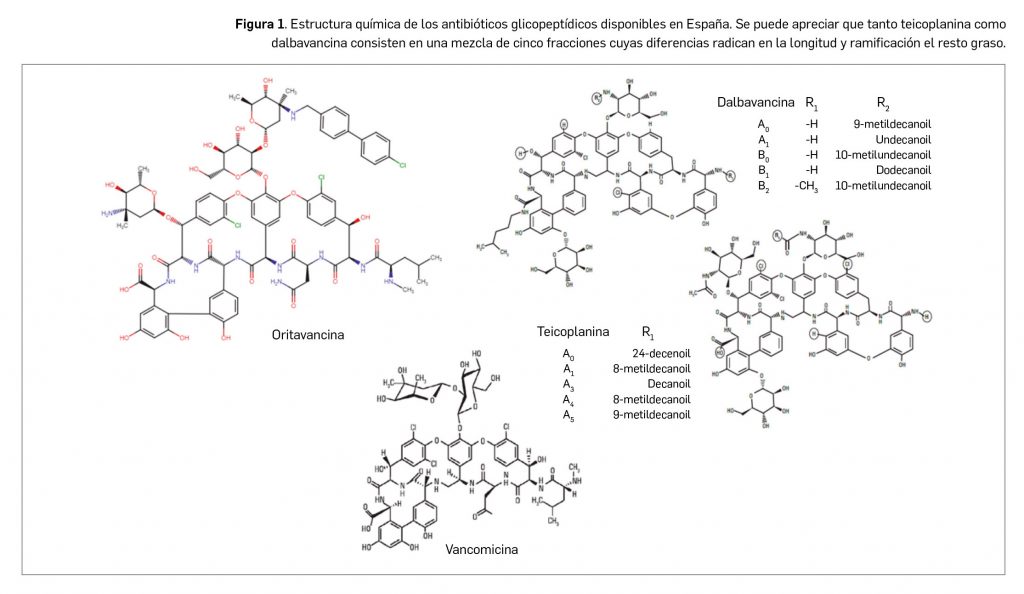
El principio activo se presenta como una sal difosfato (1:2) que se corresponde con el nombre químico [4”R]-22-O-(3-amino-2,3,6-trideoxi-3-C-metil-α-L-arabino-hexopiranosil)-N3’’-[(4’-cloro[1,1’-bifenil]-4-il)metil] vancomicina fosfato y una fórmula molecular C86H97Cl3N10O26 y un peso molecular de 17,93 KDa. Es un sólido de color blanco a rosa pálido soluble en agua y en dextrosa al 5%, cuya solubilidad decrece considerablemente hacia valores de pH neutro o básicos. No se han observado polimorfos de oritavancina, pero la molécula sí muestra estereoisomería por la presencia de 22 carbonos asimétricos y tres elementos quirales adicionales asociados a la rotación restringida de la unidad de bifenilo y de los residuos de difenil éter.
El mecanismo de acción de los antibióticos (lipo)glicopeptídicos recuerda al de los betalactámicos, pues ambos grupos actúan interfiriendo con los procesos de síntesis y reparación de la pared bacteriana, específicamente sobre el peptido¬glucano. Se ha descrito que los antibióticos (lipo)glicopeptídicos se unen de forma selectiva a las unidades de UDP-pentapéptido, que contienen un resto de D-Ala-D-Ala, mediante el grupo carboxílico libre en la molécula, con lo que provocan una acu¬mulación de los precursores UDP-N-acetilmuramilpép¬tido. La unión al UDP-pentapéptido se debe a que el antibiótico (lipo)glicopeptídico tiene una zona hidrofóbica –restos clorobencénicos unidos por puente oxi– que favorece su unión al péptido, la cual se inicia por la interacción entre el grupo carboxílico peptídico y una amina protonada del antibiótico.
A diferencia del mecanismo de acción de vancomicina, que es simple (inhibición de la transglicosilación), el mecanismo de acción de la oritavancina es triple, según se ha indicado.
Eficacia y seguridad clínicas
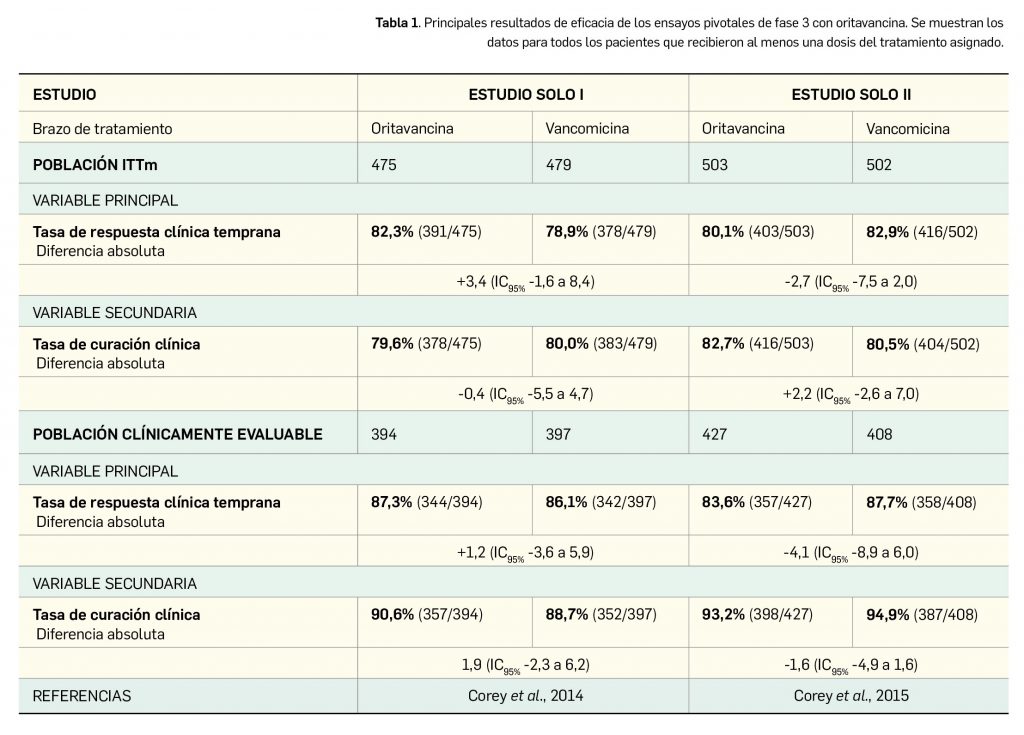
La eficacia clínica de oritavancina por vía intravenosa en la indicación y dosis autorizadas ha sido adecuadamente contrastada en dos ensayos pivotales de fase 3 –SOLO I y SOLO II– de idéntico diseño: aleatorizado (1:1), multinacional y multicéntrico, doblemente ciego, de grupos paralelos y controlado por comparador activo. Ambos estudios siguieron las guías vigentes en la UE sobre antimicrobianos a fin de investigar la no inferioridad de una dosis única del nuevo fármaco (1.200 mg) frente al uso también intravenoso de vancomicina (1 g o 15 mg/kg) dos veces al día durante 7-10 días, e incluyeron inicialmente un conjunto de 2.364 adultos con IPTBs, de los cuales 1.959 recibieron al menos una dosis de tratamiento.
Estos pacientes debían tener sospecha o confirmación de una infección bacteriana aguda y complicada de la piel y sus estructuras (ABSSSI) por bacterias grampositivas que cumpliera los criterios de la Sociedad Americana de Enfermedades Infecciosas (> 75 cm2 de enrojecimiento, induración o edema, y coexistencia de al menos un signo clínico de infección sistémica como inflamación de ganglios linfáticos, fiebre alta o leucocitosis, entre otros), bien fueran casos de celulitis/erisipelas, infecciones de heridas o abscesos cutáneos mayores. Debían requerir al menos 7 días de antibioterapia intravenosa y, en ausencia de signos de infección, tenían que ser mayores de 70 años, diabéticos o haber recibido terapia inmunosupresora o quimioterapia en los 3 meses previos. Por el contrario, se excluyeron aquellos pacientes con tratamiento antibiótico activo frente a bacterias grampositivas en las 2 semanas previas, con bacteriemia, neutropenia, contraindicación de vancomicina o no dispuestos a dejar de usar antipiréticos, entre otros.
Sus características basales estuvieron bien equilibradas en ambos estudios pivotales y entre grupos de tratamiento. Así, al inicio del estudio SOLO I la mitad de los pacientes tenía celulitis/erisipelas y un 30% padecía abscesos cutáneos mayores, oscilando el tamaño medio del área afectada entre 226 cm2 y 248 cm2 para los brazos de vancomicina y oritavancina, respectivamente; en el SOLO II se registró una tasa del 31% de casos de celulitis/erisipelas, un 32% de abscesos cutáneos mayores y un 37% de infecciones de heridas, situándose la extensión media basal de la infección entre 288 cm2 (grupo oritavancina) y 309 cm2 (grupo vancomicina). En global, se identificó el agente causal en el 37% de los casos de celulitis, en el 77% de las infecciones de heridas y en el 84% de los abscesos cutáneos; en más de la mitad de los pacientes aleatorizados a oritavancina (53%) y a vancomicina (54%) se aisló al menos un microorganismo grampositivo, de los cuales el más frecuente fue S. aureus (48%; 21% resistentes a meticilina), seguido de especies de Streptococcus (los mayoritarios fueron S. anginosus y S. pyogenes). Los casos de septicemia al inicio fueron muy escasos.
La variable principal de eficacia en ambos estudios fue la respuesta clínica temprana en la población por intención de tratar modificada (ITTm)5, evaluada a las 48-72 h tras el inicio de tratamiento y definida como una medida compuesta por: cese de la expansión o reducción del tamaño de la lesión basal, ausencia de fiebre y no necesidad de antibioterapia de rescate. Como variable secundaria –considerada primaria por la Agencia Europea de Medicamentos (EMA)– se midió la curación clínica en la visita de evaluación tras 7-14 días desde la finalización del tratamiento, tanto en la población ITTm como, dentro de ella, en la población clínicamente evaluable (pacientes tratados durante ≥ 7 días y con resultados de evaluación clínica).
Los resultados con los tratamientos antibióticos citados en la población ITTm (n= 1.959, 954 en SOLO I y 1.005 en SOLO II) se resumen en la siguiente Tabla 1 y demuestran la no inferioridad de oritavancina frente a vancomicina (fijada estadísticamente por un límite inferior del IC95% superior al -10%). Hay que subrayar que se adicionó aztreonam y metronidazol al tratamiento de los pacientes con infecciones mixtas, y que la pauta posológica de vancomicina se ajustó a las prácticas de cada centro hospitalario.
Finalmente, la seguridad del nuevo fármaco ha sido bien caracterizada por los datos de más de 3.000 sujetos que lo recibieron durante su desarrollo clínico, incluidos 1.075 pacientes con IPTBs tratados con una dosis única en los estudios de fase 2 y 3. En el conjunto de los ensayos pivotales, la incidencia global de eventos adversos –incluidos los graves y los conducentes a la interrupción del tratamiento– y de muertes fue similar para oritavancina y para vancomicina. En general, los efectos adversos relacionados con complicaciones infecciosas fueron solo ligeramente mayores con el uso de oritavancina (16% vs. 14% con vancomicina), siendo los más reportados la celulitis y los abscesos.
Entre las reacciones adversas más frecuentes a oritavancina (sobre vancomicina) destacan las náuseas (9,9%), el dolor de cabeza (7,1%), los vómitos (4,6%) y los eventos adversos hepáticos (4,7%). Otras que ocurrieron con menor frecuencia que con vancomicina fueron las reacciones de hipersensibilidad (12% vs. 19%), sobre todo prurito y urticaria.
En cualquier caso, una amplia mayoría de los eventos adversos fueron leves-moderados en severidad, destacando como reacción adversa grave más incidente la celulitis (1,1%). Si bien la tasa global de interrupción del tratamiento por motivos de seguridad fue baja (3,7% vs. 4,2% con vancomicina), la celulitis fue también la principal causa (0,4%), seguida de la osteomielitis como complicación de la infección (0,3%). Ningún caso de muerte se consideró relacionado con la administración del nuevo antibiótico. Cabe destacar que la tasa de notificación de reacciones adversas fue mayor en las mujeres que en los hombres y que, pese a la escasez de datos en adultos mayores, también parece que la tolerabilidad del fármaco es peor en mayores de 75 años.
Aspectos innovadores
Oritavancina es un nuevo antibiótico perteneciente al grupo de los lipoglicopéptidos que ejerce su efecto antibacteriano a través de un triple mecanismo de acción sobre la síntesis y reparación de la pared bacteriana (inhibe la transglicosilación mediante su unión al péptido precursor de los peptidoglicanos, inhibe la transpeptidación por su unión a los segmentos de formación de puentes peptídicos e interrumpe la integridad de la membrana bacteriana), provocando erosiones y un aumento de la permeabilidad en dicha pared que permite el contacto directo entre citoplasma bacteriano y el entorno químico externo. Ello resulta en una muerte rápida de las bacterias y ha sido la base de la aprobación del medicamento para el tratamiento, en dosis única por vía intravenosa y según las directrices oficiales del uso apropiado de antibacterianos, de pacientes adultos con infecciones bacterianas agudas de la piel y tejidos blandos de la piel (IPTBs).
Autorizado por primera vez en la UE bajo el nombre comercial de Orbactiv® (ahora se comercializa por primera vez en España), el fármaco ha probado en estudios in vitro y clínicos su actividad microbicida frente a las bacterias grampositivas más frecuentemente implicadas en las IPTBs, destacando Staphylococcus aureus (sensible o resistente a meticilina, SARM) y los estreptococos betahemolíticos (como Streptococcus pyogenes) o del grupo viridans, entre otras. Presenta, pues, un espectro de actividad similar a otros (lipo)glicopéptidos aprobados con igual indicación. No tiene actividad frente a bacterias gramnegativas ni se conoce ningún caso de resistencia cruzada con otras clases de antibióticos. Tampoco es esperable que bacterias resistentes a glicopéptidos sean sensibles a oritavancina, si bien su mecanismo más complejo puede reducir el desarrollo de resistencias.
Las IPTBs constituyen un grupo heterogéneo de enfermedades, que pueden ser agudas y graves en cualquier sujeto o, aun siendo leves, tener importancia clínica en pacientes con comorbilidades. Además del estado clínico y la gravedad de la infección, su abordaje suele considerar la epidemiología local de resistencias a antimicrobianos: junto a la cirugía o el drenaje de la lesión, la antibioterapia empírica –mayoritariamente a nivel hospitalario– debe cubrir las bacterias grampositivas más comunes y suele hacerse con penicilinas, cefalosporinas, clindamicina o cotrimoxazol si no se sospecha de SARM, recurriendo a un antibiótico que cubra dicho patógeno en caso de sospecha fundamentada, como linezolid o tedizolid, ceftarolina, daptomicina, tigeciclina o los glicopéptidos (vancomicina, teicoplanina) o lipoglicopéptidos (dalvabancina) disponibles.
Muchos de esos tratamientos implican pautas multidosis por vía intravenosa (dalvabancina solo dos dosis) y, dado que en el marco de los PROA –programas de optimización y uso racional de antibióticos– se prefiere idealmente un manejo extrahospitalario del paciente tras la fase aguda (para evitar los costes relacionados con la hospitalización y los riesgos de la vía parenteral), se está empezando a optar por pautas como el uso de vancomicina con posibilidad de cambio a terapia oral con linezolid tras el 3er día.
La aprobación de oritavancina se sustentó en dos amplios ensayos pivotales de fase 3, de adecuado y similar diseño (aleatorizados, doblemente ciego, de grupos paralelos y con comparador activo), que corroboraron su no inferioridad frente a vancomicina, precisamente un fármaco considerado de elección frente a IPTBs, de los más usados frente a infecciones por SARM. La comparabilidad de eficacia se verificó en términos de la variable primaria, esto es, la tasa de respuesta clínica temprana evaluada a las 48-72 h tras iniciar tratamiento: 80-82% con oritavancina y 79-83% con vancomicina; esas tasas aumentaban hasta el 84-87% y el 86-88%, respectivamente, si solo se consideraban los pacientes tratados durante ≥ 7 días. Igualmente, se verificó la no inferioridad para la tasa de curación clínica tras 7-14 días del fin del tratamiento (variable secundaria): fue del 80-83% con oritavancina y del 80-81% con vancomicina; en pacientes tratados más allá de una semana, se alcanzó una tasa del 91-93% y 89-95%, respectivamente. Es preciso subrayar que la EMA considera esta segunda variable como principal, de modo que ha sido la empleada para su autorización en la UE.
La evidencia disponible se ve limitada por la exclusión de los estudios pivotales de pacientes con IPTBs distintas a celulitis, abscesos e infecciones de heridas; así pues, pese a que no se puede concluir respecto a la eficacia de oritavancina frente a otros tipos de IPTBs, parece plausible extrapolar las consideraciones de no inferioridad. Tampoco se tienen datos que defiendan el uso del fármaco en infecciones por estafilococos con sensibilidad reducida a vancomicina.
No existen por ahora otras comparaciones directas de oritavancina con otros antibióticos, tampoco con los glicopéptidos semisintéticos estrechamente relacionados. Las comparaciones indirectas disponibles –de escasa robustez estadística– sugieren que las respuestas clínicas que aportan oritavancina y dalbavancina no difieren significativamente en la población global de pacientes con IPTBs (OR: 1,36; IC95% 0,85-2,18) ni en pacientes infectados por SARM (OR: 1,29; IC95% 0,11-15,48). También son parejas las respuestas clínicas comparativas entre oritavancina y telavancina (OR: 0,98; IC95% 0,72-1,31) o entre dalbavancina y telavancina (OR: 0,72; IC95% 0,45-1,13). Dalvabancina y oritavancina parecen mostrar un mejor perfil de seguridad que telavancina, y pueden considerarse como alternativas coste-efectivas a otros antibióticos usados como tratamiento estándar en la práctica clínica (Agarwal et al., 2018), con un perfil beneficio-riesgo equivalente al de vancomicina (Jame et al., 2021).
En relación con la seguridad, el perfil toxicológico de oritavancina ha sido bien caracterizado con datos de más de 3.000 sujetos tratados y parece relativamente benigno, comparable al de otros glicopéptidos. La mayoría de las reacciones adversas al fármaco se presentan con una incidencia similar al uso de vancomicina, mayor en mujeres y en pacientes de > 75 años de edad, y son leves-moderadas en severidad, con una baja tasa de interrupción del tratamiento (< 4%). Destacan por su frecuencia las reacciones de hipersensibilidad (12%, como prurito y urticaria), las náuseas (10%), el dolor de cabeza (7%), los vómitos (5%) y los eventos adversos hepáticos (5%); la celulitis es la reacción adversa grave más reportada (1%).
En definitiva, se trata de un nuevo antibiótico que se representará una alternativa más en un grupo terapéutico bien provisto, en que ya se dispone de opciones para la antibioterapia ambulatoria (linezolid, tedizolid y dalvabancina) y por vía parenteral (vancomicina, linezolid, tedizolid, daptomicina). Con estrecha relación en estructura y farmacología con dalvabancina, la elevada vida media de orivatancina (200-300 h) puede ser una ventaja de cara a la adherencia terapéutica (dosis única), por ejemplo frente a vancomicina o teicoplanina, pero a la vez favorecer la aparición de resistencias y ser un inconveniente en casos de problemas de seguridad y para conseguir el desescalado.
Así pues, el IPT establece que para el uso empírico sería más conveniente usar otras alternativas que puedan retirarse fácilmente cuando se conozca el agente causal y con las que se tiene mayor experiencia de uso (AEMPS, 2022). Pese a que oritavancina no parece implicar ninguna mejora clínica sustancial en su indicación, la incorporación de nuevos agentes antibacterianos con actividad frente a microbios multirresistentes es siempre una buena noticia para la sociedad.
Valoración