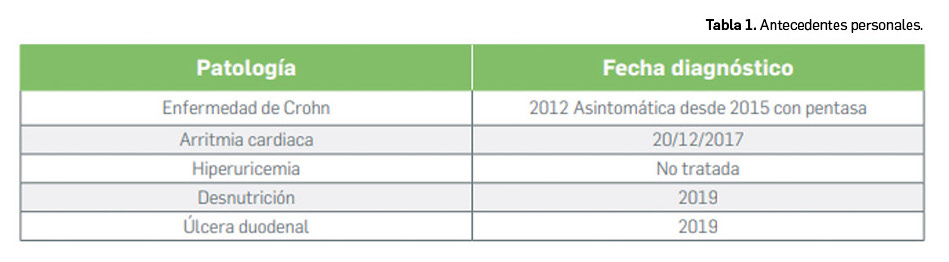
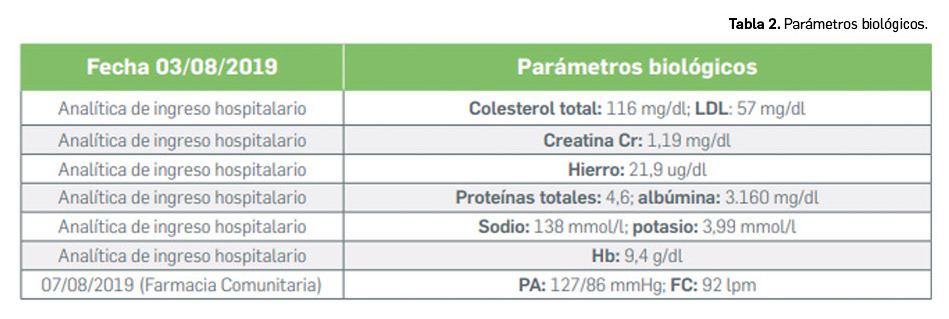
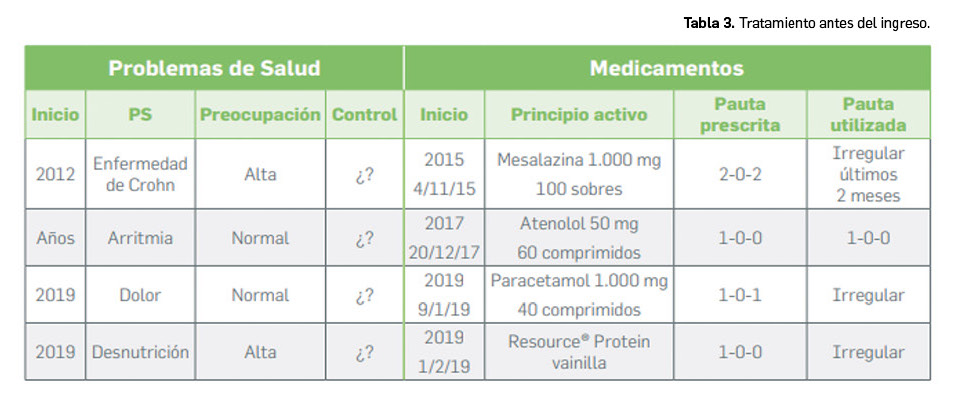
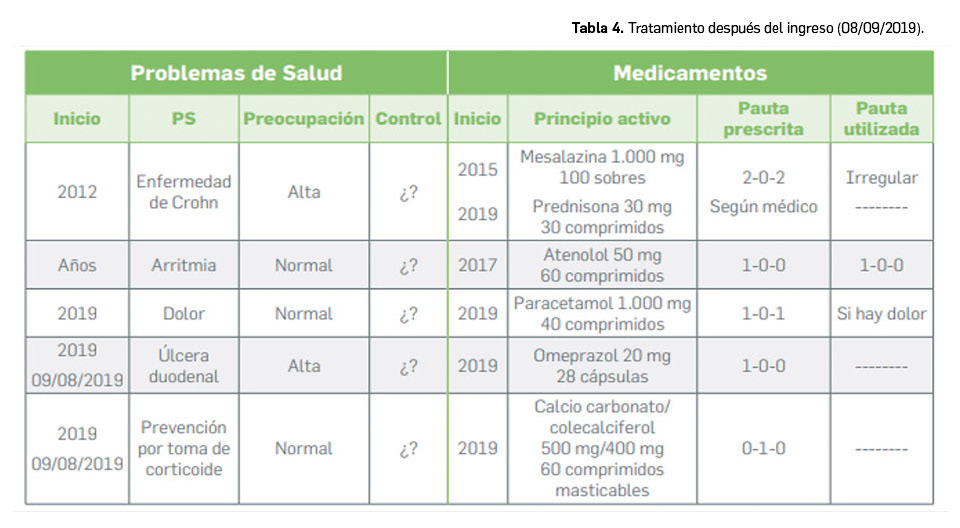
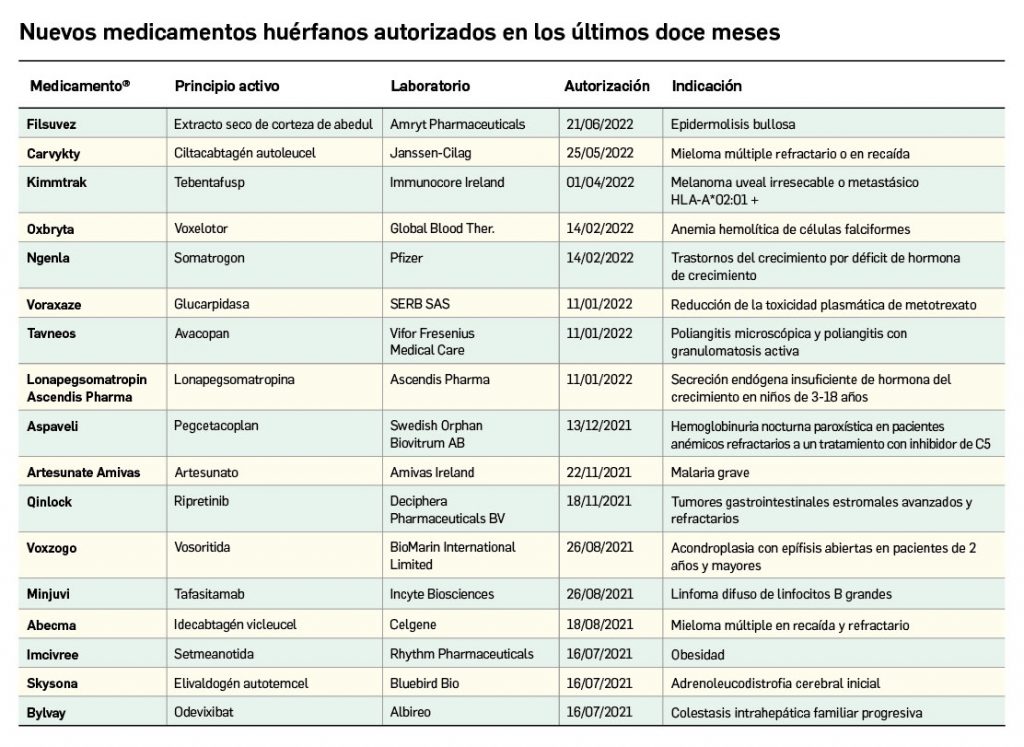
Los medicamentos huérfanos son aquéllos que sirven para diagnosticar, prevenir o tratar enfermedades raras de carácter muy grave o con riesgo para la vida. En la Unión Europea, la calificación de enfermedad rara se aplica a todas aquellas que no afectan a más de 5 de cada 10.000 personas. La designación de un medicamento como huérfano no garantiza su uso en la condición designada y no implica necesariamente que el producto satisfaga los criterios de eficacia, seguridad y calidad necesarios para la concesión de la autorización de comercialización. Como para cualquier medicamento, estos criterios solo pueden ser evaluados una vez que la solicitud de autorización de comercialización haya sido presentada.
Instituciones y redes europeas
► Agencia Europea de Medicamentos (EMA; Europea Medicines Agency). Apartado de Medicamentos Huérfanos
(inglés): https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview
https://www.ema.europa.eu/en/committees/committee-orphan-medicinal-products-comp
► Comisión Europea: web oficial de la Comisión Europea sobre enfermedades raras y medicamentos huérfanos (español).
http://ec.europa.eu/health/rare_diseases/policy/index_es.htm
► Orphanet: Portal de información oficial de la Unión Europea sobre enfermedades raras y medicamentos huérfanos (español).
http://www.orpha.net/consor/cgi-bin/index.php?lng=ES
► Eurordis: Federación Europea de Asociaciones de Pacientes con Enfermedades Raras (español). http://www.eurordis.org/es
Otras instituciones y redes internacionales
► Food & Drug Administration (FDA, Estados Unidos). Apartado de Medicamentos Huérfanos (inglés):
http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/default.htm
► Pharmaceuticals & Medical Devices Agency. Agencia de Medicamentos y Dispositivos Médicos, de Japón (inglés):
http://www.pmda.go.jp/english/index.html