Resumen
El ácido bempedoico es un nuevo profármaco que, al activarse por unión a la coenzima A, actúa como inhibidor competitivo de la enzima adenosina trifosfato-citrato liasa, de modo que inhibe la síntesis de colesterol en el hígado y reduce sus niveles intracelulares, así como los de colesterol-LDL en plasma. El medicamento ha sido autorizado para el tratamiento diario por vía oral de pacientes adultos con hipercolesterolemia primaria (familiar heterocigótica y no familiar) o dislipidemia mixta, como adyuvante a la dieta, bien en combinación con una estatina sola o con una estatina y otros tratamientos hipolipemiantes en pacientes que no puedan alcanzar sus objetivos de c-LDL con la dosis máxima tolerada de una estatina; también se ha aprobado en monoterapia o en combinación con otros tratamientos hipolipemiantes en pacientes intolerantes o con contraindicación al uso de estatinas.
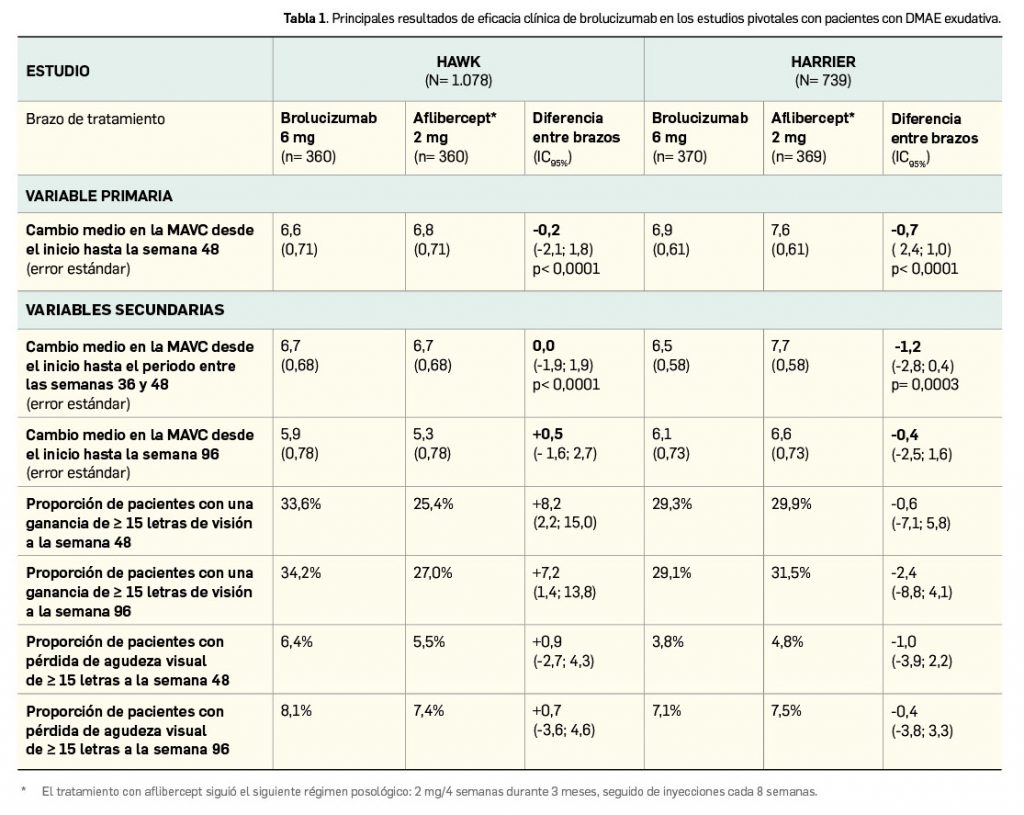
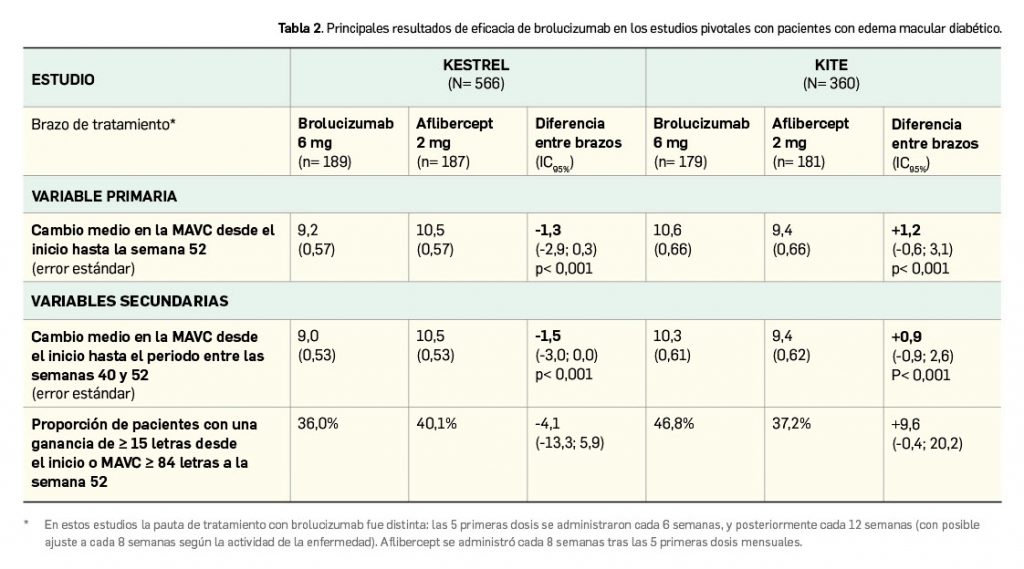
En un total de 4 ensayos pivotales de adecuado diseño (N> 3.600) ha demostrado un efecto hipolipemiante moderado. En pacientes con alto riesgo cardiovascular, su asociación a dosis máximas de estatinas redujo significativamente los niveles de c-LDL a las 12 semanas de tratamiento, en mayor medida que placebo (diferencia de -18%), y permitió a una mayor proporción de pacientes llegar al objetivo de < 70 mg/dl (29% vs. 8%); también probó su eficacia en este grupo de pacientes en combinación con dosis fijas con ezetimiba (reducción de c-LDL en -38% de diferencia con placebo). En pacientes intolerantes a las estatinas, la administración de ácido bempedoico indujo un descenso sustancial en los niveles medios de c-LDL a los 3 meses, con un -25% de diferencia respecto a placebo. Además, redujo globalmente los niveles de colesterol no HDL, de apolipoproteína B y de colesterol total, con una eficacia consistente en todos los subgrupos de pacientes, que se mostró máxima tras 1 mes y se mantuvo en niveles similares durante periodos de al menos 1 año. En términos de seguridad, se trata de un fármaco con un perfil toxicológico relativamente benigno y manejable clínicamente. La incidencia de reacciones adversas es baja y similar al uso de placebo, siendo en su mayoría leves-moderadas, por lo que la tasa de interrupción del tratamiento es también baja. Sobresalen por su frecuencia (< 5%) las siguientes reacciones adversas: hiperuricemia, mialgias, espasmo muscular, diarrea y cefalea. Se ha descrito una mayor incidencia de alteraciones hepáticas y renales, pero no parecen tener una gran relevancia clínica.
No se ha confirmado aún la eficacia del fármaco sobre la incidencia de eventos cardiovasculares y la morbi-mortalidad asociada. Ese objetivo clínico sí ha sido probado, en cambio, con el uso de estatinas, ezetimiba y los inhibidores de PCSK9 (evolocumab y arilocumab); las comparaciones indirectas con estos últimos apuntan a una inferioridad del ácido bempedoico. Tampoco se conoce su balance beneficio-riesgo en tratamientos de más de 1 año, por lo que habrá que esperar a los resultados de los estudios ahora en marcha para sacar conclusiones sólidas. En definitiva, pese a que incorpora un novedoso mecanismo de acción y es bien tolerado, el beneficio clínico esperable con el nuevo fármaco es modesto. Su adición a la terapia intensiva con estatinas y ezetimiba permitiría rescatar a un 15-20% de pacientes de alto riesgo adicionales, si los niveles de c-LDL están < 20% por encima del nivel objetivo; y en pacientes intolerantes a estatinas, su uso junto a ezetimiba podría favorecer una reducción de c-LDL un 20-30% adicional (insuficiente en la mayoría de pacientes de alto riesgo). El ácido bempedoico se posicionará, pues, como una alternativa terapéutica en tercera línea de tratamiento, sobre todo en pacientes no candidatos al uso de inhibidores de PCSK9. No parece incorporar cambios sustanciales en la terapéutica estándar de la hipercolesterolemia.
Aspectos fisiopatológicos
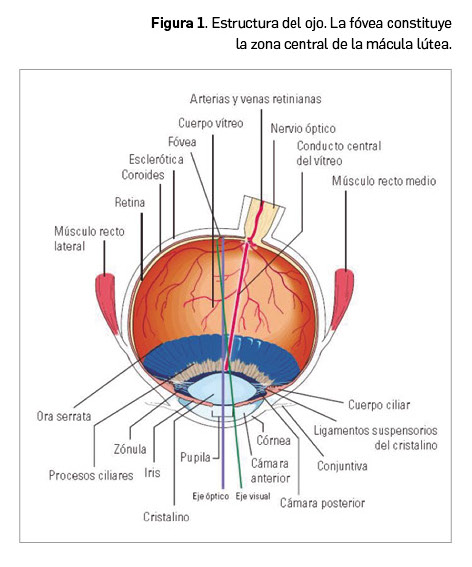
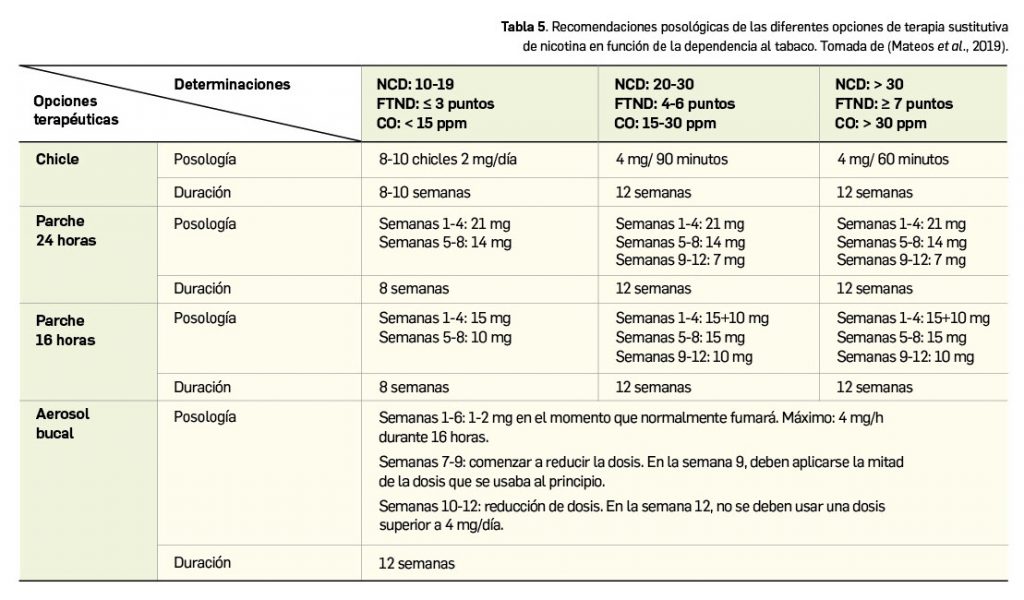
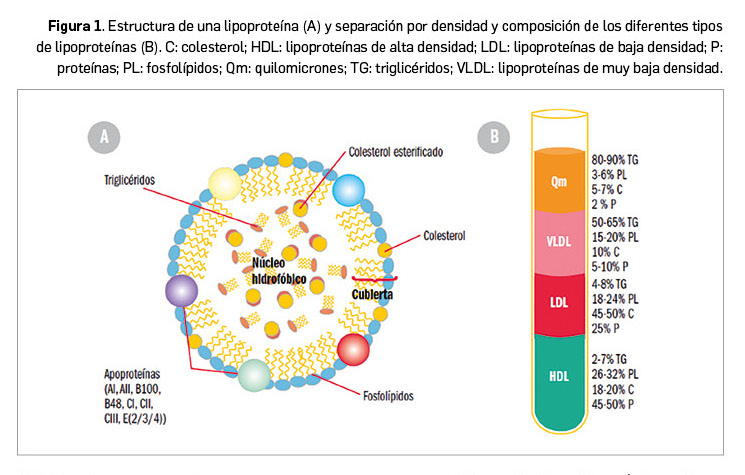
Los lípidos son sustancias que nuestro organismo emplea como almacén de energía y que desempeñan importantes funciones (formación de las membranas celulares, modificación de proteínas, producción de hormonas, etc.). Por su carácter liposoluble precisan para su transporte –en el medio acuoso que constituye el plasma– de un vehículo adecuado: colesterol, triglicéridos y fosfolípidos se transportan en el plasma formando macrocomplejos en los que las moléculas más hidrófobas se disponen en el centro y se recubren de otras hidrófilas, como fosfolípidos y apolipoproteína. Estos macrocomplejos son lo que conocemos como lipoproteínas (Figura 1), entre las que se diferencian varios tipos en función de la composición del núcleo lipófilo y del tipo de apoproteínas que presentan (algunas constituyen la tarjeta de presentación de las lipoproteínas a su receptor). Los niveles plasmáticos de lipoproteínas reflejan, por tanto, los de los lípidos que contienen.
Las lipoproteínas y el colesterol
Los ácidos grasos constituyen la base de los lípidos del organismo. Son ácidos orgánicos con cadenas hidrocarbonadas de diferente longitud y grado de saturación, que se esterifican con otras sustancias como glicerol y ácido fosfórico, dando lugar entre otros productos a triglicéridos y fosfolípidos, o bien son metabolizados a esteroles, dando lugar al colesterol. Los ácidos grasos no esterificados se asocian a la albúmina, mientras que los restantes constituyen las lipoproteínas que permiten movilizar los triglicéridos desde el intestino y el hígado hasta los puntos de utilización o almacenamiento (tejido adiposo y músculo) y transportar el colesterol allí donde se necesita: por ejemplo, para la síntesis de membranas, la producción de hormonas esteroideas o la síntesis de ácidos biliares.
Las lipoproteínas se clasifican según su densidad (a mayor densidad, mayor contenido en proteínas, y a mayor diámetro, mayor contenido de lípidos) y cada tipo ejerce distintas funciones:
- Quilomicrones: se forman en el intestino, contienen apoproteína B48 y transportan fundamentalmente los triglicéridos, los fosfolípidos y el colesterol procedentes de la dieta y de la síntesis en el epitelio intestinal. En los tejidos periféricos ricos en lipoproteína lipasa (LPL), liberan ácidos grasos.
- Lipoproteínas de muy baja densidad (VLDL, very low density lipoproteins): contienen apoproteína B (ApoB), C y E y transportan triglicéridos y colesterol, sintetizados en el hígado, hacia los tejidos periféricos, cediendo igualmente ácidos grasos en tejidos periféricos.
- Lipoproteínas de baja densidad (LDL, low density lipoproteins): proceden de las VLDL (presentan, pues, las mismas apoproteínas) y representan el principal sistema de transporte del colesterol hacia las células de los tejidos periféricos.
- Lipoproteínas de alta densidad (HDL, high density lipoproteins): sintetizadas en su mayoría en hígado e intestino, contienen fundamentalmente apoproteína A (ApoA), C y E. Transportan el colesterol cedido por las células de los tejidos periféricos al hígado. Es este transporte inverso, por tanto, un mecanismo bioquímico hipocolesterolemiante, y la razón por la que los niveles altos de c-HDL se asocian a un menor riesgo de cardiopatía isquémica.
Los enterocitos liberan los lípidos absorbidos tras la ingesta de alimentosa la linfa en forma de quilomicrones ricos en triglicéridos. En los capilares del tejido adiposo y muscular esquelético, ricos en lipoproteína lipasa (LPL), los lípidos contenidos en los quilomicrones son hidrolizados a ácidos grasos, que son asimilados por las células de estos tejidos, donde se almacenan, constituyendo un depósito de energía, o se emplean como materia prima para la síntesis de otros lípidos (Figura 2). Los quilomicrones remanentes, que no han perdido su colesterol, sufren un proceso de endocitosis en el hígado.
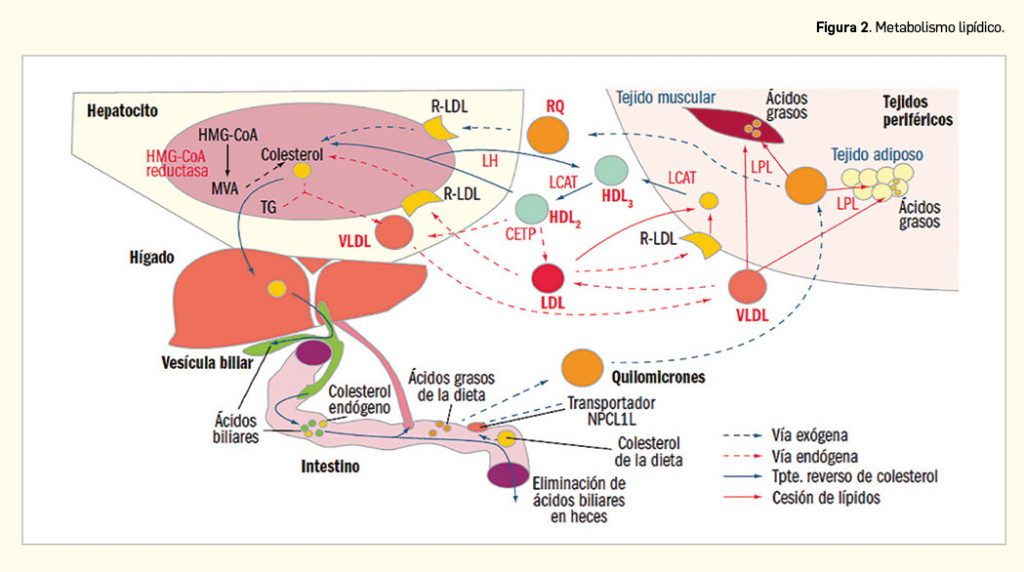
El colesterol hepático, endógeno o exógeno, puede ser esterificado y almacenado, excretarse en la bilis como colesterol libre o ser transformado en ácidos biliares, constituir nuevas lipoproteínas o incorporarse a las membranas plasmáticas de los hepatocitos. Aproximadamente el 80% del colesterol endógeno procede de síntesis hepática y se libera a la circulación en forma de VLDL; en los capilares del músculo esquelético y del tejido graso, las VLDL sufren el ataque de la LPL, que hidroliza los triglicéridos y libera ácidos grasos, que son captados por los tejidos y convierten a las VLDL en LDL. Los niveles elevados de LDL y su oxidación forman parte de la base etiopatogénica de la aterosclerosis. Las LDL circulantes son reconocidas por receptores presentes en las membranas celulares (R-LDL), produciéndose su endocitosis al interior celular, donde se convierten en sustrato de enzimas lisosomales, cediendo colesterol libre.
____________________________________________________________________________________________
Las dislipemias son alteraciones del metabolismo lipídico que conducen a desviaciones de los valores normales de las lipoproteínas o los lípidos en sangre. Aproximadamente uno de cada cuatro pacientes adultos que acuden a consultas de atención primaria o especializada presentan algún tipo de dislipemia. Se denominan primarias cuando se deben a una alteración genética o a una dieta inadecuada, y secundarias cuando están relacionadas con patologías que alteran el metabolismo lipídico1, como son la diabetes mellitus, trastornos renales, hipotiroidismo o la administración de algunos medicamentos (por ejemplo, los antirretrovirales inhibidores de la proteasa, la ciclosporina o las tiazidas).
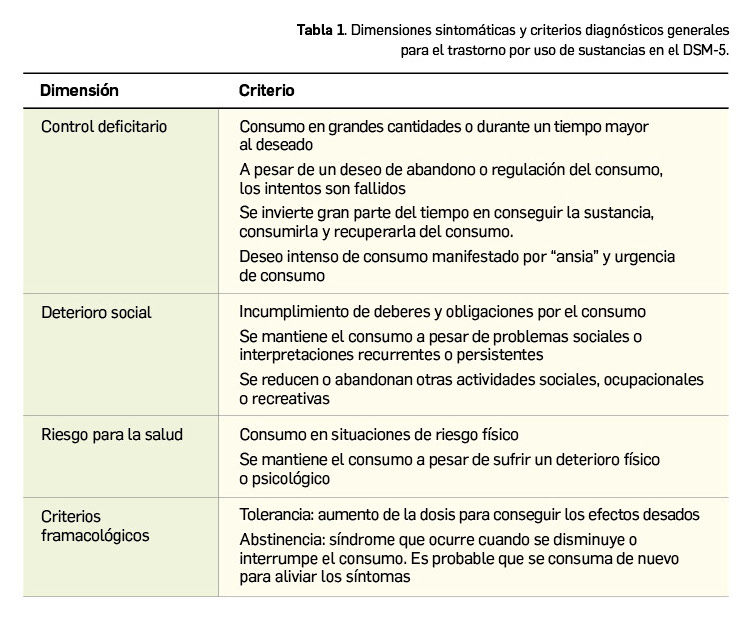
Las dislipemias han sido objeto de distintos sistemas de clasificación (Tabla 1), pero se sabe que en cada uno de los tipos se pueden encontrar distintas enfermedades (primarias o secundarias a otras patologías) que pueden dar lugar a la alteración de los niveles de un tipo de lipoproteína. La reducción del nivel del colesterol unido a LDL (en adelante, c-LDL) y los triglicéridos retrasa la progresión e incluso puede reducir la placa de ateroma. Todos los datos disponibles indican que el control de las dislipemias es esencial en la prevención primaria y secundaria de las enfermedades cardiovasculares.
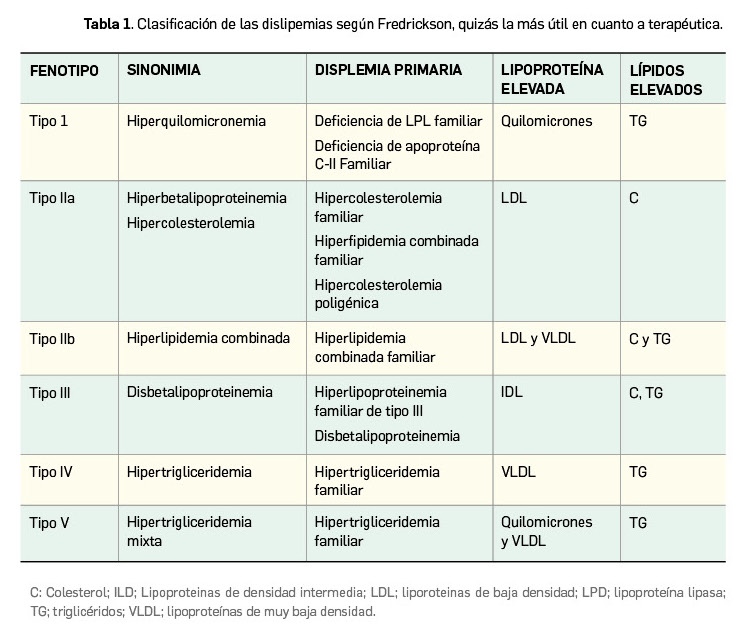
La hipercolesterolemia es la dislipemia más frecuente, presentándose bien en su forma pura (70%) o bien como dislipemias mixtas en que también se elevan los niveles de triglicéridos (25%). El incremento de los niveles de c-LDL y la disminución de los de colesterol unido a HDL (c-HDL) constituyen, junto con la hipertensión y el tabaquismo, los principales factores de riesgo modificables para el desarrollo de aterosclerosis, una enfermedad sistémica que afecta a las arterias, alterándolas progresivamente y formando placas esclerosadas que llegan a reducir la luz vascular (estenosis) y conducen a situaciones con mayor o menor grado de isquemia del área afectada y riesgo de eventos cardiovasculares.
La forma más común de hipercolesterolemia primaria es la hipercolesterolemia no familiar o poligénica (HP), que representa el 80% de los casos; en ella intervienen factores poligénicos en interacción con factores ambientales, especialmente la dieta. Por su parte, la hiperlipidemia familiar es un trastorno genético en el que se agrupan varias patologías, entre las cuales la más común es la hipercolesterolemia familiar (HF), caracterizada por la presencia de elevadas concentraciones sanguíneas de c-LDL y por el desarrollo prematuro de xantomas y enfermedades cardiovasculares. Se trata de una enfermedad hereditaria de transmisión autosómica dominante y que se manifiesta desde el nacimiento, debida a la existencia de mutaciones del gen del receptor de LDL (R-LDL) en el 95% de los casos, aunque también se asocia con mutaciones de los genes codificadores de Apolipoproteína B (ApoB) y de la PCSK9 (Proprotein convertase subtilisin/kexin type 9), un enzima que se une al R-LDL presente en los hepatocitos, facilitando su internalización y degradación.
En su forma heterocigótica, la HF tiene una prevalencia en la UE de 1:200 a 1:500 y, a pesar de un tratamiento hipolipemiante intensivo, presenta una tasa de mortalidad doble de lo normal; se estima que en España afecta a unas 100.000-190.000 personas, existiendo un enorme infradiagnóstico. La forma homocigótica es aún mucho más grave, cursando con niveles de c-LDL y colesterol total extraordinariamente elevados, lo que determina una alta mortalidad incluso en edad infantil y juvenil. Afortunadamente, es mucho más infrecuente que la forma heterocigótica, con una prevalencia de entre 1:300.000 y 1:1.000.000; se estima que en España puede afectar solo a unos 150 pacientes.
Existe abundante documentación clínica que demuestra que los niveles plasmáticos altos de colesterol se relacionan con un aumento de la incidencia de la morbimortalidad cardiovascular. Según se ha sugerido anteriormente, el principal riesgo del mantenimiento de niveles altos de lípidos en sangre se asocia con el desarrollo de la placa de ateroma y la consecuente aterosclerosis: la primera causa de muerte e incapacidad en los países desarrollados. Se considera éste como un proceso inmunoinflamatorio crónico de origen multigénico y multifactorial, que comienza tempranamente por una disfunción del endotelio vascular y modificaciones oxidativas de los lípidos sanguíneos atrapados en el subendotelio. Los lípidos depositados en las lesiones ateroscleróticas provienen fundamentalmente de las LDL circulantes, que ingresan en la pared vascular a través de este endotelio lesionado o disfuncional. La enfermedad se desarrolla de forma intermitente en el tiempo, observándose un crecimiento discontinuo de las placas (se intercalan periodos de inactividad con otros de rápida evolución), modulado por factores ambientales y genéticos. El resultado final es que aparecen y se desarrollan lesiones con mayor o menor contenido lipídico, presencia de colágeno, fibras elásticas y calcio, que, en casos avanzados, pueden sufrir complicaciones. La localización y el grado de desarrollo de estas lesiones determinan las diferentes formas clínicas, con manifestaciones correspondientes a la disminución del flujo y aparición de isquemia crónica, o bien a la obstrucción de dicha luz por rotura/fisura de la placa y aparición de fenómenos trombóticos (el núcleo lipídico de la placa desencadena la activación de las cascadas de la coagulación y de la adhesión y agregación plaquetarias) e isquemia aguda.
La prevención y tratamiento de las dislipemias debe contemplarse como parte fundamental de la prevención de la patología cardiovascular (la primera causa de muerte en todo el mundo, según la OMS). Como tal, sus objetivos deben ajustarse atendiendo al riesgo cardiovascular que presenta el paciente concreto. A lo largo de numerosos y amplios ensayos clínicos, se ha evidenciado que la reducción de los niveles séricos de colesterol total y de c-LDL comporta una reducción del riesgo cardiovascular, por lo que esto se convierte en objetivo prioritario del tratamiento, al que pueden seguir los valores de otras fracciones lipídicas asociadas a la aterosclerosis. Por ello, el criterio principal subrogado para evaluar la eficacia del tratamiento2–reducción del riesgo cardiovascular en sus diversas manifestaciones patológicas (número de eventos de infarto de miocardio, accidente cerebrovascular, etc.)– es el descenso de la fracción LDL y el aumento de la fracción HDL.
Los pilares del abordaje de las hipercolesterolemias son la dieta, el ejercicio y la terapia farmacológica hipolipemiante. La justificación del tratamiento hipolipemiante en todos los casos de pacientes con hipercolesterolemia es aún hoy objeto de polémica y está lejos de haberse demostrado esta máxima terapéutica para todos los fármacos disponibles: hay una tendencia a no considerar solo los niveles de c-LDL, evitándose la farmacoterapia hipocolesterolemiante en pacientes jóvenes sin factores de riesgo cardiovascular; en cambio, se suele optar por intensificarlos en pacientes que hayan sufrido un episodio coronario o presenten, en general, un nivel de riesgo coronario y/o cerebrovascular de moderado a elevado.
En general, con cifras de disminución del c-LDL del orden del 25-30% hay un retardo significativo en la progresión de la arteriosclerosis en arteria coronaria y carotídea (la regresión de las lesiones se produce pocas veces). Clásicamente, se han considerado como objetivos terapéuticos los siguientes niveles plasmáticos: colesterol total < 200 mg/dl, colesterol-LDL < 100 mg/dl, colesterol-HDL > 60 mg/dl y triglicéridos < 150 mg/dl. Algunas guías clínicas fijan objetivos del tratamiento en función de la presencia o no de enfermedad coronaria y de la existencia de factores adicionales de riesgo. En todo caso, la terapia hipolipemiante parece evolucionar a tratamientos cada vez más selectivos, pero también más intensivos. Se dispone actualmente de distintas estrategias farmacoterapéuticas que se definen a continuación. Los denominados inhibidores de la HMG-CoA reductasa, más comúnmente referidos como estatinas, constituyen el tratamiento de elección de las hipercolesterolemias. Actúan inhibiendo competitivamente uno de los enzimas claves en el proceso de síntesis del colesterol en el organismo: la HMG-CoA reductasa. La inhibición de esta enzima produce una activación de las proteínas reguladoras SREBP (sterol regulatory elements-binding proteins), que activan a su vez la transcripción de proteínas e incrementan la expresión del gen del receptor de LDL y un aumento en la cantidad de receptores funcionales en el hepatocito, dando lugar a una mayor eliminación de las LDL circulantes, lo que disminuye las concentraciones de c-LDL. El tratamiento con estatinas produce una reducción rápida e intensa de colesterol total (15-30%) y de c-LDL (25-60%), un descenso moderado de triglicéridos (10-25%; de hasta 35% en sujetos con niveles de > 250 mg/dl) y un ligero ascenso (4-10%) de c-HDL. El orden de potencia hipolipemiante a nivel equimolecular es: pitavastatina > rosuvastatina > atorvastatina > simvastatina > pravastatina = lovastatina > fluvastatina; sin embargo, cuando se utilizan dosis equivalentes, el efecto terapéutico es similar. Al doblar la dosis de cualquier estatina, el aumento relativo en la reducción de c-LDL es, en general, del 6-7%.
Estos fármacos actúan rápidamente, inhibiendo la enzima limitante en pocas horas; asimismo, más del 90% del efecto reductor de c-LDL se consigue las primeras 4 semanas de tratamiento, sin que sea haya evidenciado tolerancia farmacológica con el tratamiento crónico. Además de sus efectos sobre el perfil lipídico, las estatinas tienen otros efectos cardiovasculares beneficiosos, especialmente sobre la pared arterial, que explicarían el beneficio adicional no atribuible a la reducción del c-LDL observado en muchos estudios (efectos pleiotrópicos). Al inhibir la HMG-CoA reductasa, interfieren en la formación de isoprenoides a partir del mevalonato, y reducen la prenilación de las proteínas G (Rho, Rac, Rab y Ras) necesaria para su anclaje a la membrana celular y su correcto funcionamiento. A través de estos potenciales efectos, las estatinas pueden inhibir el crecimiento de la célula muscular lisa, la adhesión celular, la activación plaquetaria y la secreción de proteína C reactiva.
Hay una amplia experiencia de la buena tolerabilidad con el uso de estos fármacos, aunque ocasionalmente producen un aumento de las transaminasas séricas (que revierten al suspender el tratamiento) o miositis. No obstante, un porcentaje significativo de pacientes (se estima en 10-15%) presentan intolerancia a estatinas, principalmente por mialgia acompañada de elevaciones significativas de creatina kinasa (CK), que pueden desembocar en rabdomiolisis en los casos más severos.
De igual modo, muchos pacientes no alcanzan el nivel objetivo de c-LDL a pesar del tratamiento intensivo. Antes de cualquier intensificación del tratamiento, las guías clínicas recomiendan reevaluar la adherencia del paciente a la farmacoterapia y a los cambios en el estilo de vida (dieta, ejercicio). En caso de que sea necesario intensificar el tratamiento, la aféresis de LDL es un tratamiento a considerar, pero presenta limitaciones prácticas, y es más común recurrir a otras alternativas farmacológicas, como las siguientes:
- Los fibratos son derivados del ácido clofíbrico que reducen fundamentalmente los niveles de triglicéridos (30-50%), y consecuentemente, los de c-VLDL, y aumentan los de c-HDL (10-20%). La acción reductora sobre colesterol total (10-20%) y c-LDL (5-10%) es comparativamente mucho menor, y variable según el fármaco. Los únicos que permanecen en el mercado son: fenofibrato, bezafibrato y gemfibrozilo. Este último es un poco distinto: tiene escaso efecto sobre el colesterol, pero una acción más pronunciada sobre triglicéridos, y en ciertos casos produce una elevación significativa de la fracción c-HDL.
- Los secuestrantes de sales biliares –colesevelam y colestiramina– son polímeros de intercambio aniónico que constituyen el grupo de hipolipemiantes más antiguo, aunque con una eficacia probada. Reducen los niveles de c-LDL en un 10-30% y en combinación con estatinas pueden alcanzar el 60%. Actúan uniéndose a los ácidos biliares presentes en la luz intestinal, formando complejos no absorbibles que son eliminados con las heces y, por tanto, impiden la reabsorción de dichos ácidos biliares. A medida que la reserva orgánica de estos se agota, el enzima colesterol 7α hidrolasa experimenta una regulación al alza, incrementado la tasa de conversión de colesterol en ácidos biliares, lo que a su vez provoca un aumento de la demanda de colesterol por parte de los hepatocitos, dando lugar a un incremento tanto de la expresión como de la actividad de la HMG-Coa reductasa, así como del número de receptores hepáticos de LDL. Estos efectos compensatorios potencian la eliminación de c-LDL de la sangre y, por consiguiente, reducen sus niveles. Su principal inconveniente es una elevada tasa de discontinuidad en el tratamiento, debido fundamentalmente a las molestias gastrointestinales; sobre todo, el estreñimiento, que alcanza hasta el 40% de los pacientes.
- El interés por los aceites poliinsaturados de pescado surgió de estudios epidemiológicos que sugerían que poblaciones con dietas ricas en pescado (esquimales, japoneses) tienen una incidencia de enfermedades cardiovasculares significativamente inferior a otros grupos de población con consumo equivalente de grasas animales o vegetales. Los preparados de aceite de pescado son mezclas de los ácidos eicosapentaenoico y docosaexaenoico, en que la acción farmacológica principal la ejerce el primero; el segundo actúa fundamentalmente de reserva transformándose lentamente en el primero, si bien puede tener funciones biológicas aún no bien conocidas. A dosis terapéuticas ambos se comportan como hipolipemiantes, pero también desarrollan otros efectos preventivos cardiovasculares cuya naturaleza es compleja: modifican la proporción de los diversos tipos de prostaglandinas del organismo, lo que puede traducirse en acción vasodilatadora e inhibición de la agregación plaquetaria.
- La ezetimiba es otro agente hipolipemiante, que actúa situándose en las microvellosidades del intestino delgado, donde inhibe la captación del colesterol por los enterocitos a través de la inhibición del transportador de esteroles Niemann-Pick C1-Like 1 (NPC1L1), que actúa en la absorción de colesterol y fitoesteroles. Reduce la absorción intestinal del colesterol procedente de la dieta y de la secreción biliar en más de un 50%. En términos generales, reduce los niveles de c-LDL en un 17-20% en monoterapia, y en más del 40% en combinación con estatinas; tiene una acción reductora moderada sobre triglicéridos (10%) e incrementa modestamente los niveles de HDL (5-10%).
- La incorporación más reciente en el ámbito del tratamiento de la hipercolesterolemia o enfermedad vascular establecida (cardiopatía isquémica, enfermedad cerebrovascular isquémica y enfermedad arterial periférica) han sido los inhibidores de la PCSK9 evolocumab y alirocumab. Se trata de anticuerpos monoclonales con propiedades hipolipemiantes que actúan mediante su unión a la citada proteína PCSK9 y provocan una reducción de la degradación intracelular de los receptores de c-LDL y, en consecuencia, una reducción marcada de los niveles de c-LDL en sangre. Se emplean fundamentalmente en pacientes que tienen niveles de c-LDL fuera de los objetivos terapéuticos (> 100 mg/dl) a pesar de un tratamiento intensivo con estatinas, o en caso de intolerancia o contraindicación a estatinas. Suelen ser eficaces y seguros en la gran mayoría de pacientes tratados, asociándose a posibles reacciones en el lugar de la inyección subcutánea.
Acción y mecanismo
El ácido bempedoico es un nuevo inhibidor de la enzima adenosina trifosfato-citrato liasa (ACL), que reduce el colesterol de lipoproteínas de baja densidad (C-LDL) mediante la inhibición de la síntesis de colesterol en el hígado. El medicamento ha sido autorizado para el tratamiento por vía oral de pacientes adultos con hipercolesterolemia primaria (familiar heterocigótica y no familiar) o dislipidemia mixta, como adyuvante a la dieta, bien en combinación con una estatina sola o con una estatina y otros tratamientos hipolipemiantes en pacientes que no puedan alcanzar sus objetivos de c-LDL con la dosis máxima tolerada de una estatina; también se ha aprobado en monoterapia o en combinación con otros tratamientos para la reducción de los lípidos en pacientes intolerantes a las estatinas o en los que esté contraindicada una estatina.
La enzima ACL está situada por encima de HMG-CoA reductasa (3-hidroxi-3-metil-glutarilcoenzima A reductasa) en la ruta de biosíntesis hepática del colesterol, y se encarga normalmente de la transformación de citrato en acetil coenzima A. En realidad, el ácido bempedoico es un profármaco que requiere la activación previa al unirse con la coenzima A mediante la acil-CoA sintetasa 1 de cadena muy larga (ACSVL1) expresada principalmente en hígado (no en músculo esquelético), dando lugar a su metabolito activo que inhibe de forma competitiva y potente (CI50 < 3 µM) la ACL. Se entiende que, al inhibir ese enzima, el efecto farmacológico del ácido bempedoico es la reducción de la síntesis hepática de colesterol y de sus niveles intracelulares, así como la disminución de las concentraciones plasmáticas de c-LDL mediante la regulación al alza del número de receptores de LDL en la superficie celular. Adicionalmente, se ha probado que también suprime la síntesis hepática de ácidos grasos.
Los estudios preclínicos y clínicos han permitido demostrar que el uso en monoterapia de ácido bempedoico y también asociado a otros fármacos modificadores de lípidos consigue reducir los niveles de c-LDL, colesterol no asociado a HDL, apolipoproteína B y del colesterol total en pacientes con hipercolesterolemia o dislipidemia mixta, incluyendo aquellos que presentan concomitantemente diabetes mellitus, en quienes se observó también un interesante efecto antidiabético (niveles menores de hemoglobina glicosilada HbA1c en comparación con placebo) que no aparecía en pacientes no diabéticos.
Aspectos moleculares
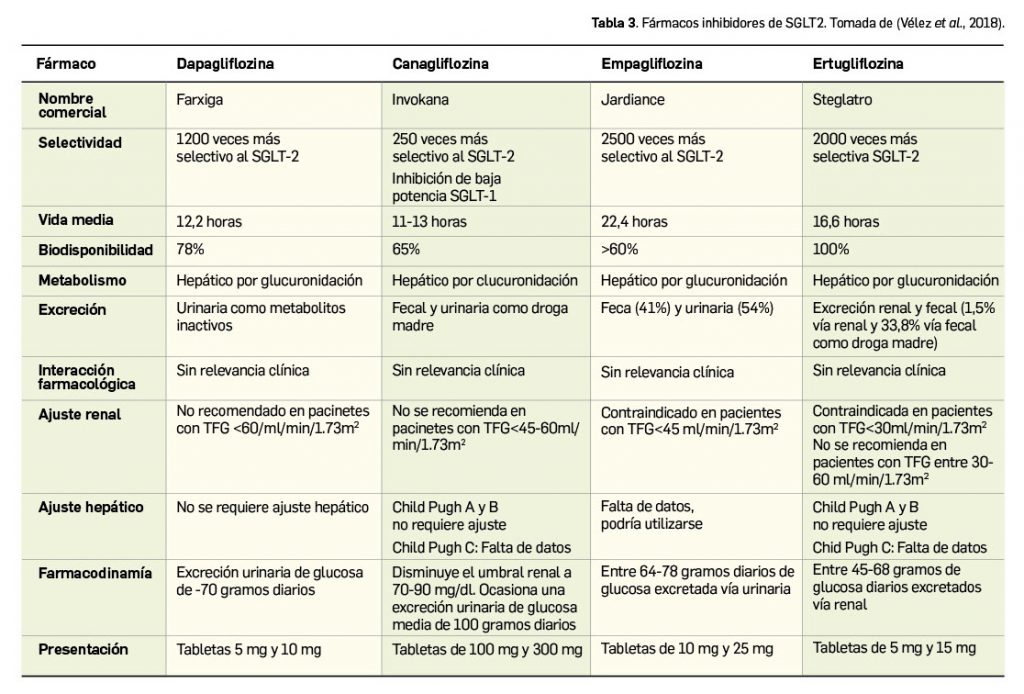
El ácido bempedoico es una molécula pequeña activa por vía oral que tiene por nombre químico el de ácido 8-hidroxi-2,2,14,14-tetrametilpentadecanedioico, que se corresponde con la fórmula molecular C19H36O5 y un peso molecular de 345 g/mol o Da (Figura 3).
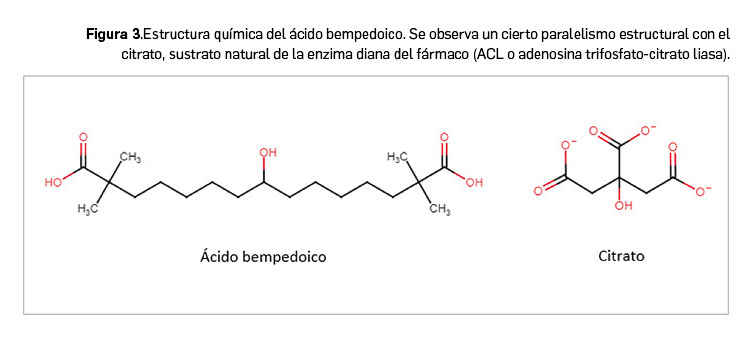
El fármaco se presenta como un polvo cristalino de color blanco o blanquecino. La molécula no tiene centros quirales ni polimorfismos. Su solubilidad es dependiente de pH: tiene una solubilidad creciente conforme aumenta el pH en el rango fisiológico normal; es insoluble a valores de pH bajos y aumenta sensiblemente su solubilidad por encima de pH= 6.
Eficacia y seguridad clínicas
La eficacia y seguridad clínicas del ácido bempedoico por vía oral han sido adecuadamente contrastadas en cuatro ensayos pivotales de fase 3 (estudios CLEAR) y similar diseño entre sí: multinacionales y multicéntricos, aleatorizados (2:1), de dos grupos paralelos, doblemente ciegos y controlados con placebo. En conjunto incluyeron a un total de 3.623 pacientes adultos con hipercolesterolemia o dislipidemia mixta (2.425 recibieron ácido bempedoico 180 mg/día y el resto placebo), que tenían un nivel de c-LDL ≥ 70 mg/dl bien el inicio. Si bien tuvieron periodos de duración diferentes, en todos ellos la variable principal de eficacia fue la reducción promedio de los niveles de c-LDL basal a las 12 semanas.
Dos de los estudios –CLEAR Harmony (Ray et al., 2019) y CLEAR Wisdom (Goldberg et al., 2019)– evaluaron el uso del fármaco en coadyuvancia a la terapia hipolipemiante, pues incluyeron pacientes con alto riesgo cardiovascular3 que estaban en tratamiento con las dosis máximas toleradas de estatinas (en monoterapia o asociadas a otros agentes hipolipemiantes), mientras que los otros dos estudios –CLEAR Serenity (Laufs et al., 2019) y CLEAR Tranquility (Ballantyne et al., 2018)– enrolaron mayoritariamente pacientes no tratados con estatinas por intolerancia4 documentada a estas.
Las principales características de los estudios, así como las particularidades demográficas y clínicas basales de los pacientes y los principales resultados de eficacia se resumen en la Tabla 2. En general, se excluyeron pacientes tratados con dosis de simvastatina > 40 mg/día, gemfibrozilo o con inhibidores de PCSK9.
De modo interesante, se debe subrayar que en los cuatro estudios el efecto del ácido bempedoico sobre la reducción de los niveles de c-LDL fue consistente en los distintos subgrupos de pacientes evaluados, confirmando su superioridad sobre placebo con independencia de factores como edad, raza, género, IMC, categoría de riesgo cardiovascular, intensidad de la terapia con estatinas, uso concomitante de otros agentes hipolipemiantes (ezetimiba o fibratos) o nivel basal de c-LDL. La mayor intensidad del efecto hipolipemiante del nuevo fármaco se vio en la semana 4 y se mantuvo posteriormente en todos los ensayos. Además, del estudio CLEAR Serenity se desprende que el fármaco es eficaz en la reducción de los niveles de c-LDL en el subgrupo de pacientes que no reciben tratamiento hipolipemiante de base (133 pacientes en el brazo experimental y 67 en el de placebo), en quienes se verifica un cambio porcentual medio de -22,1% frente a placebo tras 3 meses de tratamiento.
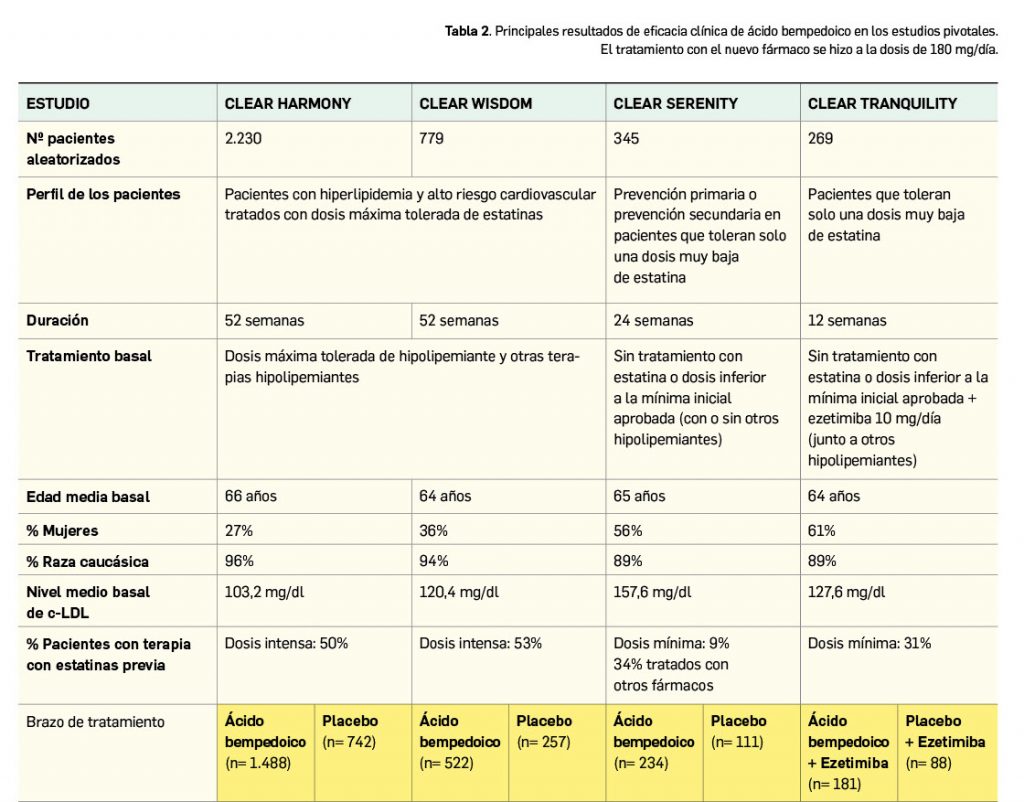
Continúa la tabla aquí
Es preciso recordar que el ácido bempedoico se ha autorizado también en otro medicamento multicomponente en asociación con ezetimiba a dosis fija (Nustendi®). El estudio pivotal conducente a su autorización fue multicéntrico, doble ciego, de 12 semanas de duración y con 4 ramas paralelas. Aleatorizó (2:2:2:1) a un total de 382 pacientes con alto riesgo cardiovascular e hiperlipidemia, si bien el análisis de eficacia incluyó solo los datos de 301 pacientes. Los brazos de tratamiento oral diario fueron: ácido bempedoico/ezetimiba 180/10 mg (n= 86), ácido bempedoico 180 mg (n=88), ezetimiba 10 mg (n=86) o placebo (n=41). Se excluyeron pacientes tratados con simvastatina a ≥ 40 mg/día, con fibratos o con inhibidores de PCSK9. Entre las características basales de los pacientes, bien balanceadas entre los brazos de tratamiento, destacan las siguientes: edad media de 64 años, 50% mujeres, 81% de raza caucásica, el nivel medio de c-LDL fue de 149,7 mg/dl, el 61-69% de los pacientes estaba en tratamiento con estatinas y el 29-41% recibía tratamiento intensivo con dichos fármacos.
Los resultados (Ballantyne et al., 2019) pusieron de manifiesto que la asociación de ácido bempedoico y ezetimiba redujo notablemente el nivel de c-LDL en la semana 12 comparado con placebo (-38%; IC95%: -46,5 a -29,6%; p< 0,001). Esa reducción también fue significativa cuando se comparó con la monoterapia con ácido bempedoico (-19%; IC95% -26,1 a -11,9; p< 0,001) y con ezetimiba (-25%; IC95% -19,7 a -6,5; p< 0,001). De nuevo, se observó el efecto máximo en la reducción de c-LDL con la combinación de fármacos en la semana 4, manteniéndose la eficacia hasta el final del estudio. Un 28% de los pacientes tratados con ácido bempedoico/ezetimiba alcanzó el objetivo terapéutico (c-LDL < 70 mg/dl) en la semana 12, frente a tan solo el 9,7%, 7,8% y 1,9% en los brazos de ezetimiba, ácido bempedoico y placebo, respectivamente. También se vieron reducciones significativas con el nuevo tratamiento en los niveles de colesterol no-HDL (-31,9% de cambio medio vs. -19,9%, -14,1% y +1,8%, respectivamente), de apolipoproteína B (-24,6% de cambio medio vs. -15,3%, -11,8% y +5,5%, respectivamente) y de colesterol total (-26,4% de cambio medio vs. -16,0%, -12,1% y +0,7%, respectivamente).
Con respecto a seguridad, el perfil toxicológico del ácido bempedoico ha sido bien definido en base a los datos de más de 3.200 pacientes con hiperlipidemia tratados diariamente con el fármaco en los 4 estudios pivotales, bien junto con estatinas a la dosis máxima tolerada (la mayoría) o bien solo con dosis mínimas o sin ese tratamiento concomitante; en el primer caso la mediana de exposición fue de 1 año, y en el segundo caso, de aproximadamente 3 meses. Los eventos adversos relacionados con el tratamiento tuvieron una incidencia muy similar a la descrita con placebo (22-25% vs. 14-22%), siendo en su gran mayoría leves-moderados en severidad (< 1% fueron graves). Las reacciones adversas descritas con mayor frecuencia (todas ellas ≤ 5%) con el uso de ácido bempedoico fueron: hiperuricemia5 (5% vs. 1,5% con placebo; gota en 1,4% vs. 0,4% de los pacientes), mialgias, espasmo muscular6 , diarrea y cefalea. Las alteraciones hepáticas –elevación de transaminasas– y alteraciones renales –fallo renal o aumento de creatinina sérica– tuvieron una incidencia ligeramente superior con el fármaco (2,8% vs. 1,3% en ambos casos), mientras que no hubo diferencias significativas entre tratamiento en cuanto al número de eventos cardiovasculares mayores.
En general, la tasa de interrupciones y suspensiones del tratamiento fue baja y también similar entre los tratados con ácido bempedoico y con placebo; en la mayoría de los casos, los motivos fueron espasmos musculares (0,7%), diarrea (0,5%), dolor en extremidades (0,4%) y náuseas (0,2%). No se han observado diferencias globales de seguridad entre adultos jóvenes y aquellos mayores de 65 años (AEMPS, 2020a; AEMPS, 2020b). Por último, también se confirmó el perfil de seguridad en su asociación con ezetimiba a dosis fija, en cuyo caso las reacciones adversas más comúnmente notificadas (< 5%) fueron hiperuricemia y estreñimiento.
Aspectos innovadores
El ácido bempedoico es un nuevo profármaco inhibidor competitivo de la enzima adenosina trifosfato-citrato liasa (enzima implicada en la ruta de biosíntesis hepática de colesterol, por encima de la HMG-CoA reductasa) que reduce los niveles de colesterol de lipoproteínas de baja densidad (n-LDL) mediante la disminución de la síntesis de colesterol en el hígado y de sus niveles intracelulares; también reduce las concentraciones plasmáticas de c-LDL mediante la regulación al alza del número de receptores de LDL en la superficie celular. Requiere ser activado previamente mediante la unión a la coenzima A, dando lugar a su metabolito activo. El medicamento ha sido autorizado para el tratamiento diario por vía oral de pacientes adultos con hipercolesterolemia primaria (familiar heterocigótica y no familiar) o dislipidemia mixta, como adyuvante a la dieta, bien en combinación con una estatina sola o con una estatina y otros tratamientos hipolipemiantes en pacientes que no puedan alcanzar sus objetivos de c-LDL con la dosis máxima tolerada de una estatina; también se ha aprobado en monoterapia o en combinación con otros tratamientos para la reducción de los lípidos en pacientes intolerantes o con contraindicación al uso de estatinas.
Su autorización se basó en 4 estudios de fase 3 de adecuado diseño (controlado por placebo, doble ciego y multicéntrico) que incluyeron a más de 3.600 pacientes y en los que demostró un efecto hipolipemiante moderado. En el conjunto de pacientes con alto riesgo cardiovascular, su asociación a dosis máximas de estatinas demostró reducciones significativamente superiores en los niveles de c-LDL a las 12 semanas de tratamiento, con una disminución de entre el 17 y 18% en comparación con placebo; además, permitió que una mayor proporción de pacientes alcanzara el objetivo recomendado de c-LDL < 70 mg/dl (29% vs. 8%). Hallazgos más interesantes se tuvieron en un estudio que investigó la combinación a dosis fijas de ácido bempedoico y ezetimiba en pacientes de alto riesgo (reducción de c-LDL de -38% en comparación con placebo). Por otra parte, en el grupo de pacientes intolerantes a las estatinas, la administración de ácido bempedoico también indujo, frente a placebo, un mayor descenso en los niveles medios de c-LDL a los 3 meses de tratamiento, aproximadamente del 25% (rango -21% a -28%); un análisis post-hoc reveló que la proporción de pacientes adicionales de alto riesgo cardiovascular que alcanzó el objetivo terapéutico fue bastante modesta (≈8%). Su efecto fue mayor en el estudio en que se administró junto con ezetimiba, lo cual sugiere un sinergismo entre ambos.
En líneas generales, el ácido bempedoico también redujo los niveles de colesterol no HDL, de apolipoproteína B y de colesterol total. Se vio que su eficacia es máxima a la semana 4 de tratamiento y se mantiene posteriormente en niveles similares durante periodos de al menos 1 año, con independencia de factores como la edad, el grado de riesgo cardiovascular, la intensidad de la dosis de estatinas, el uso concomitante de otros fármacos o el nivel basal de hiperlipidemia. Con relación a la seguridad, su perfil toxicológico, bien definido, parece relativamente benigno y manejable clínicamente. La incidencia de reacciones adversas es baja y en niveles similares a placebo, siendo en su mayoría leves-moderadas, por lo que la tasa de interrupción del tratamiento es también baja. Sobresalen por su frecuencia (< 5%) las siguientes reacciones adversas: hiperuricemia, mialgias, espasmo muscular, diarrea y cefalea. Se ha descrito una mayor incidencia de alteraciones hepáticas y renales, aunque no parecen tener una gran relevancia clínica. La principal limitación de la evidencia disponible se refiere a que, en su desarrollo clínico, el ácido bempedoico ha probado su efecto hipolipemiante en la reducción de c-LDL, pero esta es solo una variable subrogada que, aunque ha mostrado correlación con el riesgo cardiovascular, no permite confirmar el efecto del fármaco en la morbimortalidad cardiovascular. Este sería el objetivo clínico fundamental, que sí ha sido probado y cuantificado con estatinas, ezetimiba e inhibidores de PCSK9. Serán los tratamientos de duración de > 1 año en los estudios en marcha los que permitan dilucidar si el ácido bempedoico aporta un beneficio relevante.
Junto a esto, la comparación directa de ácido bempedoico únicamente frente a placebo en los estudios pivotales complica aún más su posicionamiento en el arsenal terapéutico. Además, los datos de su uso combinado con inhibidores de PCSK9 son muy limitados. Tanto evolocumab como alirocumab han evidenciado reducciones en torno al 60% en los niveles de c-LDL en estudios previos (con proporciones de pacientes que bajaban de 70 mg/dl de hasta el 74-80%), por lo que las comparaciones indirectas apuntarían a una inferioridad del ácido bempedoico. Los datos del estudio con el medicamento multicomponente con ezetimiba a dosis fijas sugieren incluso que la monoterapia con ezetimiba puede ser superior al uso de ácido bempedoico, aunque esto tampoco ha sido sólidamente probado.
Se debe recordar que en la actualidad la práctica clínica general consiste en evaluar de forma individualizada la necesidad de tratamiento hipolipemiante según riesgo cardiovascular y niveles basales y objetivos de c-LDL. Es común la adición de ezetimiba al tratamiento con la dosis máxima tolerada de estatinas en pacientes con hiperlipidemia y alto riesgo cardiovascular, lo cual reduce considerablemente los candidatos a otros tratamientos. Los inhibidores de PCSK9 son una alternativa en pacientes con hipercolesterolemia familiar o patología vascular que se mantienen fuera del objetivo terapéutico recomendado (hay debate en torno al límite de c-LDL, que oscila entre < 55 mg/dl y < 160 mg/dl). Tanto ezetimiba como los inhibidores de PCSK9 se emplearán también en caso de intolerancia o contraindicación a estatinas.
En tal contexto terapéutico, el ácido bempedoico incorpora un novedoso mecanismo de acción en la indicación, si bien, pese a que su perfil de seguridad es benigno, su eficacia parece modesta. Su adición a la terapia intensiva con estatinas y ezetimiba permitiría rescatar a un 15-20% de pacientes de alto riesgo adicionales, particularmente si los niveles de c-LDL están < 20% por encima del nivel objetivo (esa sería la reducción máxima esperable con el nuevo fármaco). En pacientes intolerantes a estatinas y riesgo intermedio-alto, su uso junto a ezetimiba podría favorecer una reducción de c-LDL un 20-30% adicional, que sería insuficiente en la mayoría de los pacientes de alto riesgo. Para alcanzar mayores reducciones, las guías clínicas establecen que se deberá seguir recurriendo, si se puede, al tratamiento con inhibidores de PCSK9.
En definitiva, el ácido bempedoico se posicionará como una alternativa terapéutica en una tercera línea de tratamiento, sobre todo en pacientes no candidatos al tratamiento con inhibidores de PCSK9. Se necesitan datos clínicos más robustos sobre su uso a largo plazo y su efecto sobre los eventos cardiovasculares y la morbi-mortalidad asociada para esclarecer su balance beneficio-riesgo, pero no parece que vaya a incorporar cambios sustanciales en la terapéutica estándar de los pacientes con hipercolesterolemia (AEMPS, 2022). No ha sido financiado para su uso en el Sistema Nacional de Salud español, por lo que el acceso efectivo a este nuevo tratamiento será limitado.
Valoración