Resumen
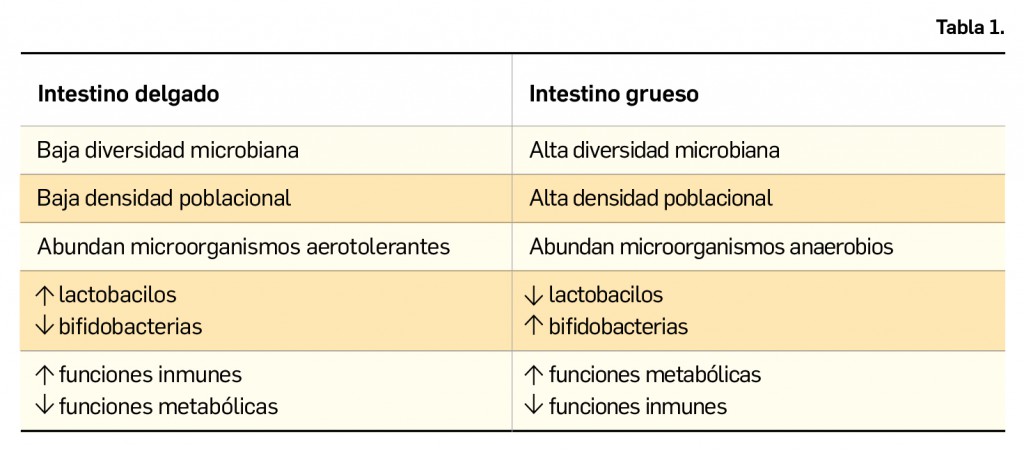
BOT PLUS identifica, a través de unos pictogramas concretos, aquellos medicamentos con problemas de suministros oficiales publicados en la página web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). Además, con el nuevo buscador de problemas de suministro, el farmacéutico podrá conocer de forma rápida si un determinado medicamento se encuentra afectado por dichos problemas u obtener listados de medicamentos, lo cual le permitirá mejorar la gestión de la oficina de farmacia y la atención farmacéutica.
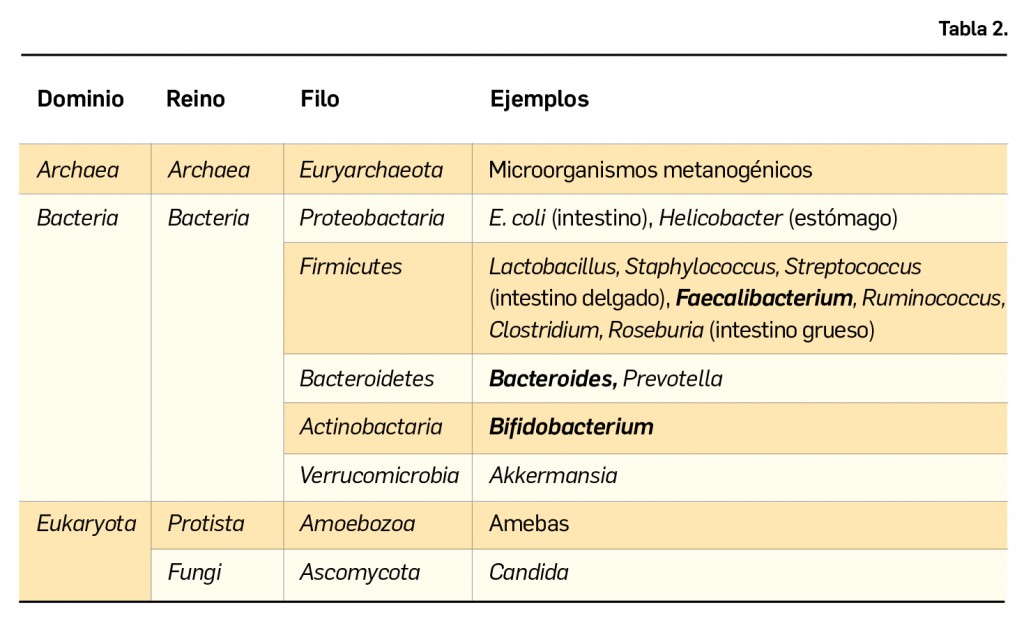
El Consejo General de Colegios Farmacéuticos utiliza la información publicada por la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) como una de las principales fuentes oficiales de información para la base de datos de información sobre medicamentos y productos de parafarmacia BOT PLUS. En el caso de la información sobre problemas de suministro oficiales publicados por la AEMPS, BOT PLUS recoge dicha información en la ficha de cada medicamento.
En BOT PLUS esta información se actualiza y codifica diariamente, de forma que los medicamentos afectados por problemas de suministro incluyen un mensaje de advertencia que permite identificarlos (Figura 1). Igualmente, se incluye un mensaje específico para identificar a aquellos medicamentos para los que la AEMPS informa de la existencia de un problema de suministro y que se puede solicitar como medicamento extranjero.

Igualmente, y para visualizar de forma rápida si un medicamento estuviera afectado por un problema de suministro, se han incluido los siguientes pictogramas en la ficha de los medicamentos para identificar ambas situaciones (Figura 2):


Para obtener más información, en los medicamentos afectados, aparece una pestaña específica sobre “Problemas de suministro” en la que se puede ampliar la siguiente información, en base a lo publicado por la AEMPS (Figura 3):
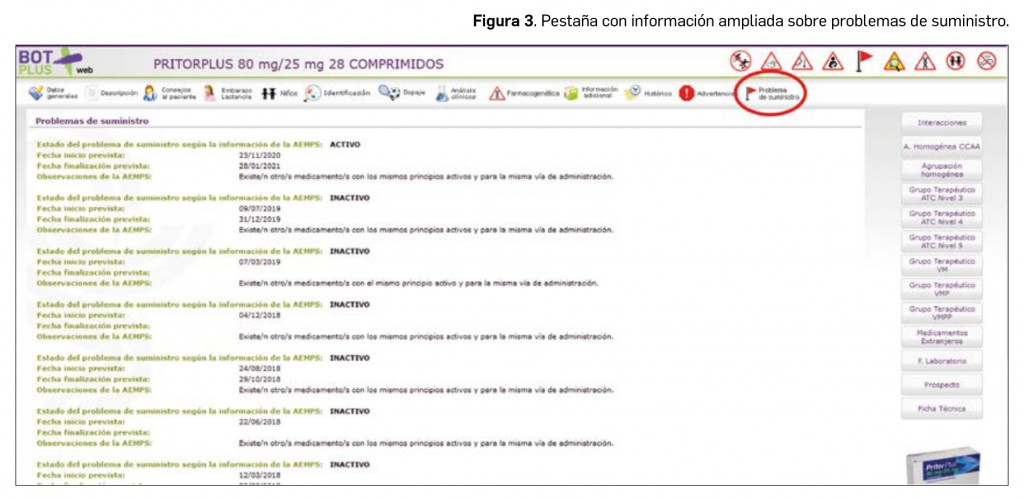
- Estado del problema de suministro según la información de la AEMPS: activo o resuelto.
- Fechas de inicio y fin previstas por el laboratorio titular, cuando éste las ha comunicado a la AEMPS.
- Observaciones de la AEMPS. Ofrece información sobre el tipo de problema de suministro y las posibles medidas que los profesionales pudieran llevar a cabo para paliarlo, tales como: “Desabastecimiento temporal”; “Se puede solicitar como medicamento extranjero”; “Existen otros tratamientos con el mismo principio activo y para la misma vía de administración”; “El titular de autorización de la comercialización está realizando una distribución controlada al existir unidades limitadas” y, en el caso de no existir sustitutos para el medicamento afectado, “El médico prescriptor deberá determinar la posibilidad de utilizar otros tratamientos”.
- Documentos relacionados: cuando ha sido publicada, se adjunta la Nota informativa de la AEMPS.
Además, para facilitar al farmacéutico la identificación de estos medicamentos, se ha creado un buscador específico de problemas de suministro. Para realizar esta búsqueda, accedemos al menú “BOT” situado en la parte superior izquierda de la pantalla principal. Una vez dentro, accedemos a “LISTADOS” y a continuación a “MEDICAMENTOS DE USO HUMANO” y seleccionamos por último “CON PROBLEMAS DE SUMINISTRO” donde se encuentra ubicado este buscador (Figura 4).
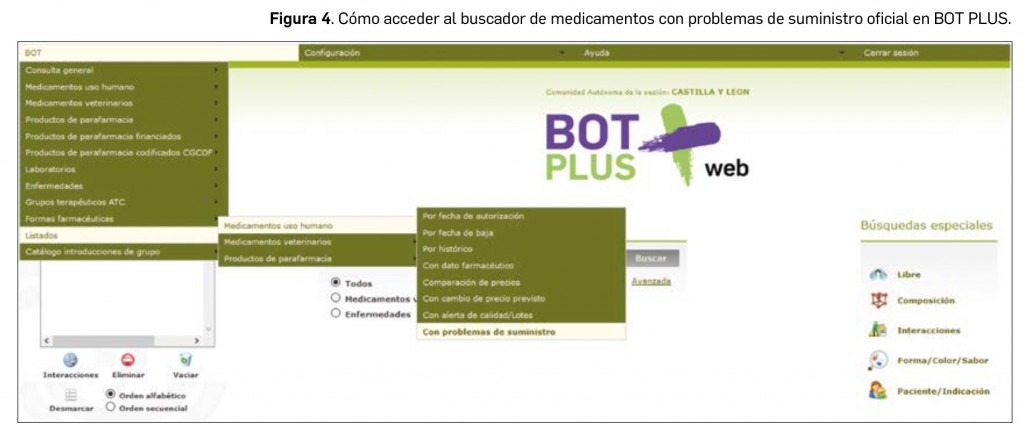
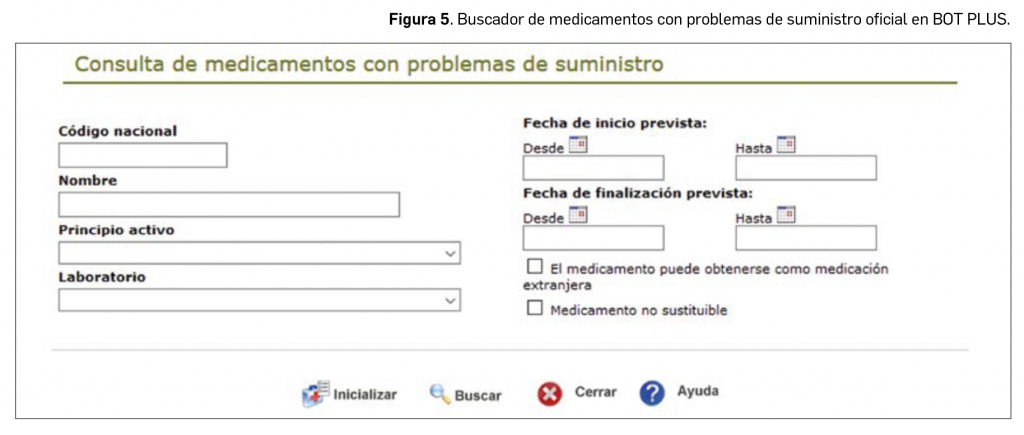
Este buscador nos permite localizar todos los medicamentos existentes con dichos problemas de suministro, así como añadir diferentes criterios para acotar la búsqueda, por ejemplo, el código nacional (si queremos consultar un medicamento concreto), buscar por principio activo, por laboratorio o filtrar los medicamentos no sustituibles (Figura 5).
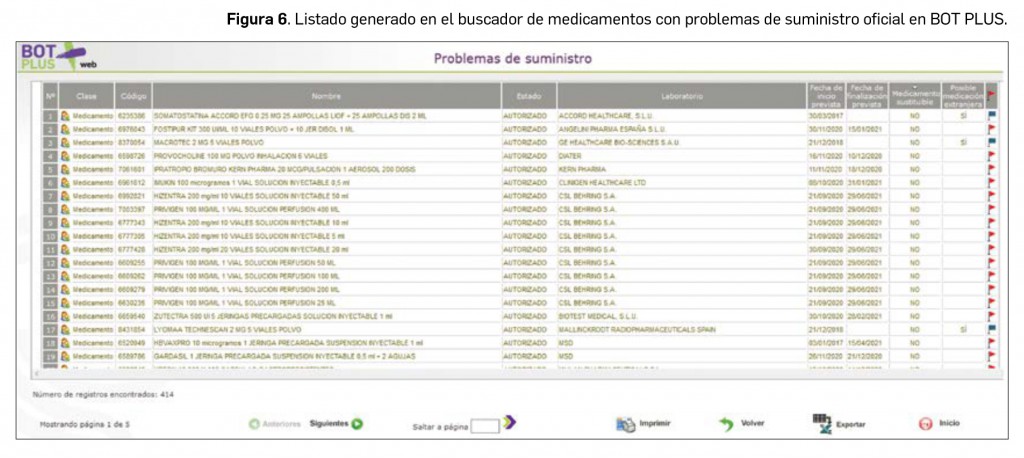
Una vez seleccionados los criterios de búsqueda, clicaremos sobre el botón de “Buscar” y se genera un listado de los medicamentos con problemas de suministro, en el cual aparecen varias columnas (fechas de inicio y fin, medicamentos no sustituibles y medicamentos extranjeros), que se pueden ordenar. Además, como todos los listados generados en BOT PLUS, también lo podemos imprimir o exportar (Figura 6).
Todos estos datos permanecen constantemente actualizados. Adicionalmente, en el Consejo General se realiza una revisión diaria de esta información para poder extraer aquellos casos que pueden plantear más dudas en la oficina de farmacia por carecer de sustitutos, información que se traslada puntualmente a todos los Colegios Oficiales de Farmacéuticos. Por otro lado, se publica mensualmente en la sección de Alertas y comunicaciones de la AEMPS de Panorama Actual del Medicamento (PAM) un listado de nuevos problemas de suministro y otro con problemas de suministro ya finalizados, hasta la fecha de cierre de cada número.
En resumen, la identificación de estos medicamentos y la posibilidad de encontrar esta información con un buscador específico en BOT PLUS supone una importante herramienta para el desempeño diario del farmacéutico.