Número 439, Diciembre 2020
_______________________________________________________________________________________________
Valoración de la innovación terapéutica en PAM
Es importante indicar que se valora el grado de innovación. Todos los medicamentos, sean innovadores o no, tienen utilidad terapéutica, en tanto que su autorización por las autoridades sanitarias implica que han demostrado rigurosamente su eficacia, su seguridad, su calidad y las condiciones de uso (incluyendo la información contenida en la ficha técnica – sumario de características – y en el prospecto del medicamento). Por tanto, la valoración que se hace se refiere a la incorporación, en el grado que se determine, de algún elemento innovador con respecto a otros medicamentos autorizados previamente para iguales o similares indicaciones terapéuticas o, en su caso, cubriendo la ausencia de éstas.
Asimismo, debe considerarse que ésta es una evaluación que se practica coincidiendo con la comercialización inicial del medicamento. Se trata, por consiguiente, de una valoración provisional de la innovación realizada en función de la evidencia clínica disponible hasta el momento, lo que no prejuzga, en ningún caso, la disponibilidad posterior de nuevas evidencias científicas (de eficacia o de seguridad) en la indicación autorizada o el potencial desarrollo y autorización, en su caso, de nuevas indicaciones terapéuticas o la imposición de restricciones de uso en las anteriores.
Se consideran tres posibles niveles, adjudicados en función de la relevancia de la(s) innovación(es) presentes en el nuevo medicamento, siempre en relación al arsenal terapéutico disponible clínicamente en España en el momento de la comercialización:
Se distinguen dos niveles de evidencia científica para los aspectos innovadores de los nuevos medicamentos:
El rigor de los datos contrastados mediante ensayos clínicos controlados (evidencia clínica) es determinante en la valoración de la innovación, mientras que las potencialidades solo pueden ser valoradas accesoriamente, como aspectos complementarios de esta valoración. En ningún caso, un medicamento es valorado con un nivel de innovación importante en función de sus ventajas potenciales, si no aporta otras ventajas demostradas clínicamente. Se analizan cinco aspectos de la innovación: clínica, molecular, toxicológica, físico-química y económico-tecnológica. Como ya se ha indicado, la fundamental y determinante es la novedad clínica.
El pasado día 13 de noviembre de 2020 se celebró por teleconferencia la reunión de trabajo del Jurado para la concesión de los Premios Panorama 2020, formado en esta ocasión por los siguientes miembros:
Los Premios Panorama valoran el grado de innovación. Conviene recordar que todos los medicamentos, tanto los no innovadores como los innovadores, tienen utilidad terapéutica en tanto que su autorización por las autoridades sanitarias (AEMPS/EMA) implica que han demostrado rigurosamente su eficacia, su seguridad, su calidad y las condiciones de uso (incluyendo la información contenida en la ficha técnica –sumario de características–
y en el prospecto del medicamento). Por tanto, la valoración que se hace en el informe previo a la deliberación del Jurado se refiere a la incorporación, en el grado indicado, de algún elemento innovador con respecto a otros medicamentos autorizados previamente para iguales o similares indicaciones terapéuticas o, en su caso, cubriendo la ausencia de éstas.
Asimismo, debe considerarse que ésta es una evaluación que se practica coincidiendo con la comercialización inicial del medicamento. Se trata, por consiguiente, de una valoración provisional de la innovación, realizada en función de la evidencia clínica disponible hasta ese momento; lo cual no prejuzga, en ningún caso, la disponibilidad futura de nuevos datos clínicos o la posible autorización de nuevas indicaciones terapéuticas, así como tampoco la potencial apari-ción de aspectos desfavorables previamente desconocidos (efectos adversos graves, contraindi-caciones, interacciones, etc.).
Se consideran tres posibles niveles, adjudicados en función en de la naturaleza de la(s) in-novación(es) presentes en el nuevo medicamento, siempre en relación al arsenal terapéutico disponible comercialmente en España en el momento de la comercialización:
Se distinguen dos niveles de evidencia científica para los aspectos innovadores de los nuevos medicamentos:
El rigor de los datos contrastados mediante ensayos clínicos controlados (evidencia clínica) sobre el grado de mejora de los resultados de la intervención con el nuevo medicamento en relación con la terapia estándar, es determinante en la valoración global de la innovación, mientras que las potencialidades solo tienen un carácter accesorio en esta valoración. En ningún caso, un medicamento es valorado con un nivel de innovación importante en función de sus ventajas potenciales, si no aporta otras ventajas demostradas clínicamente. Se analizan cinco aspectos de la innovación: clínica, molecular, toxicológica, físico-química y económico-tecnológica, aunque, como ya se ha indicado, la fundamental y determinante es la novedad clínica.
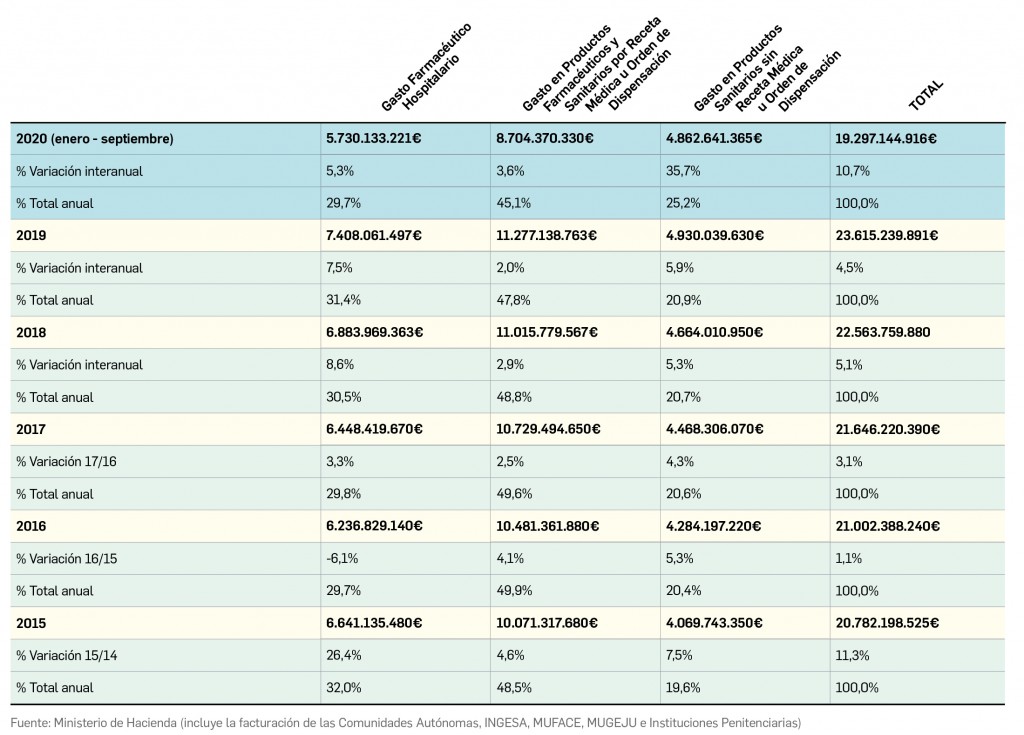
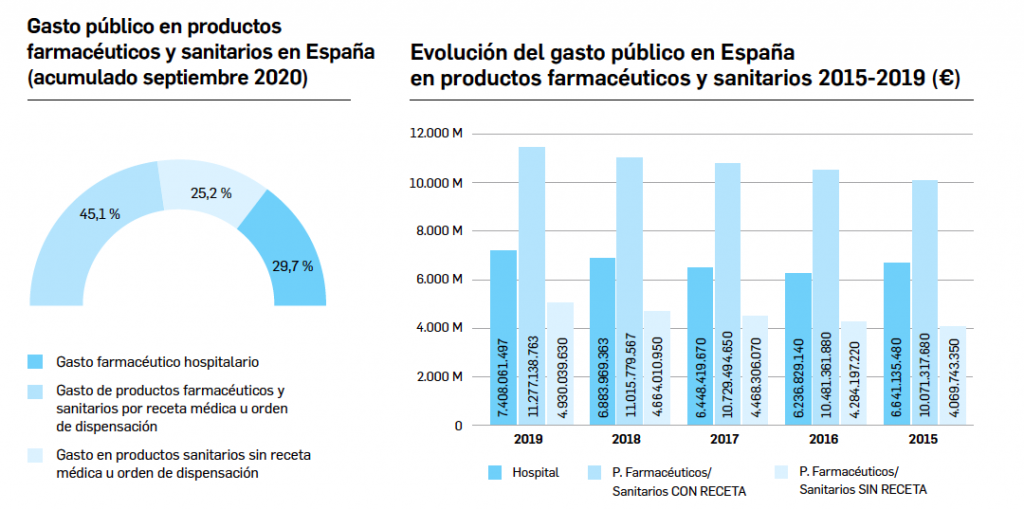
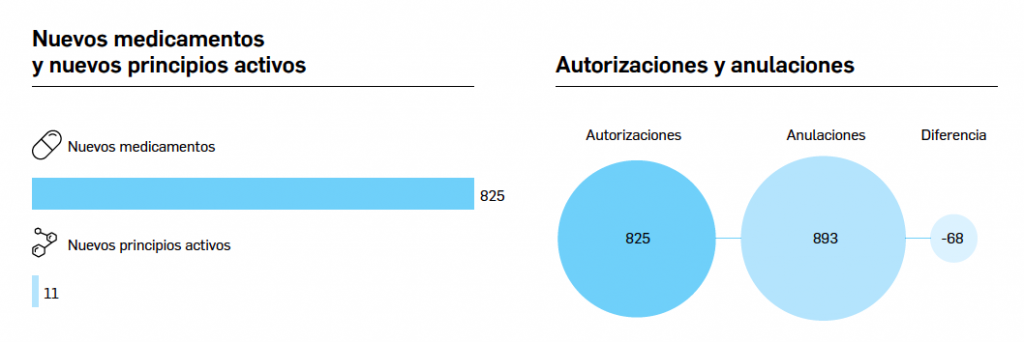
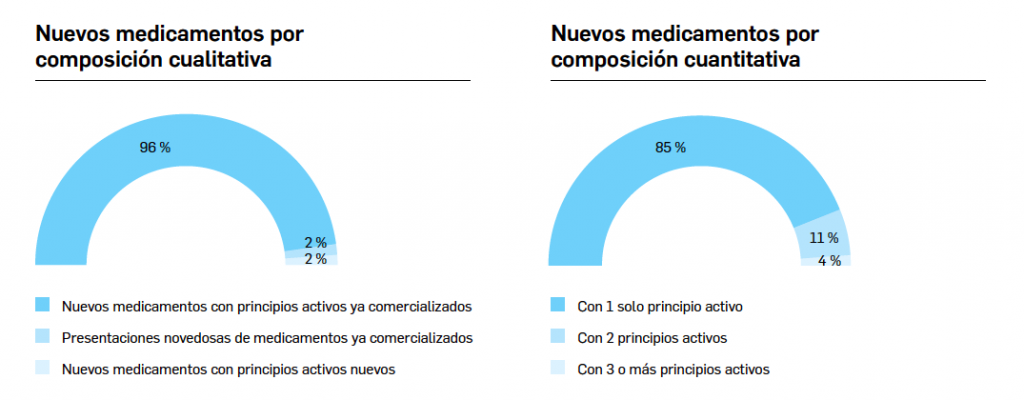
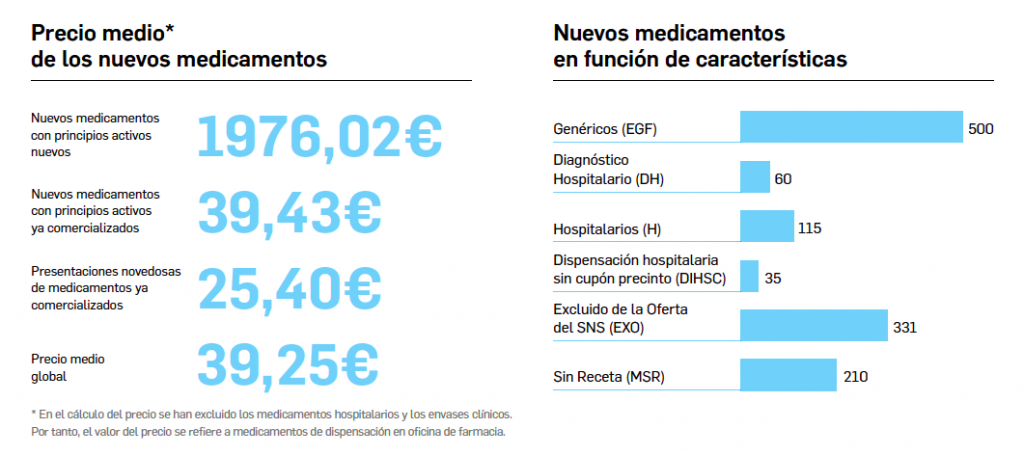
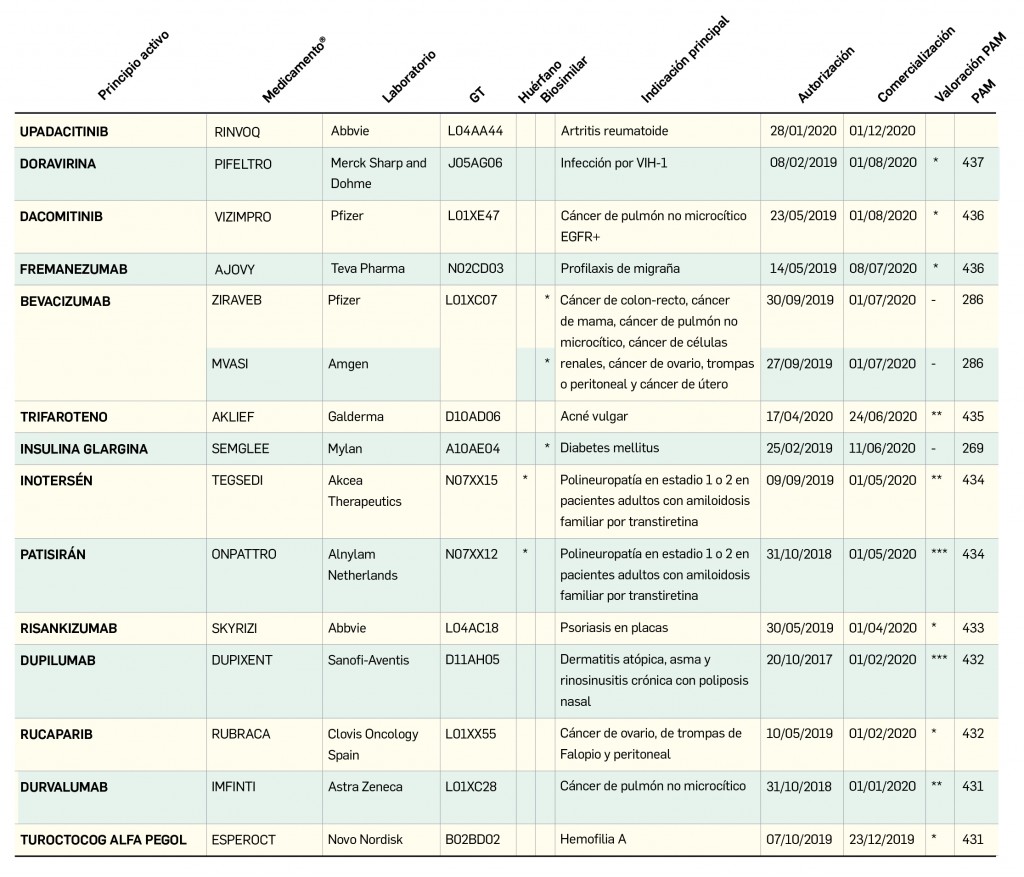
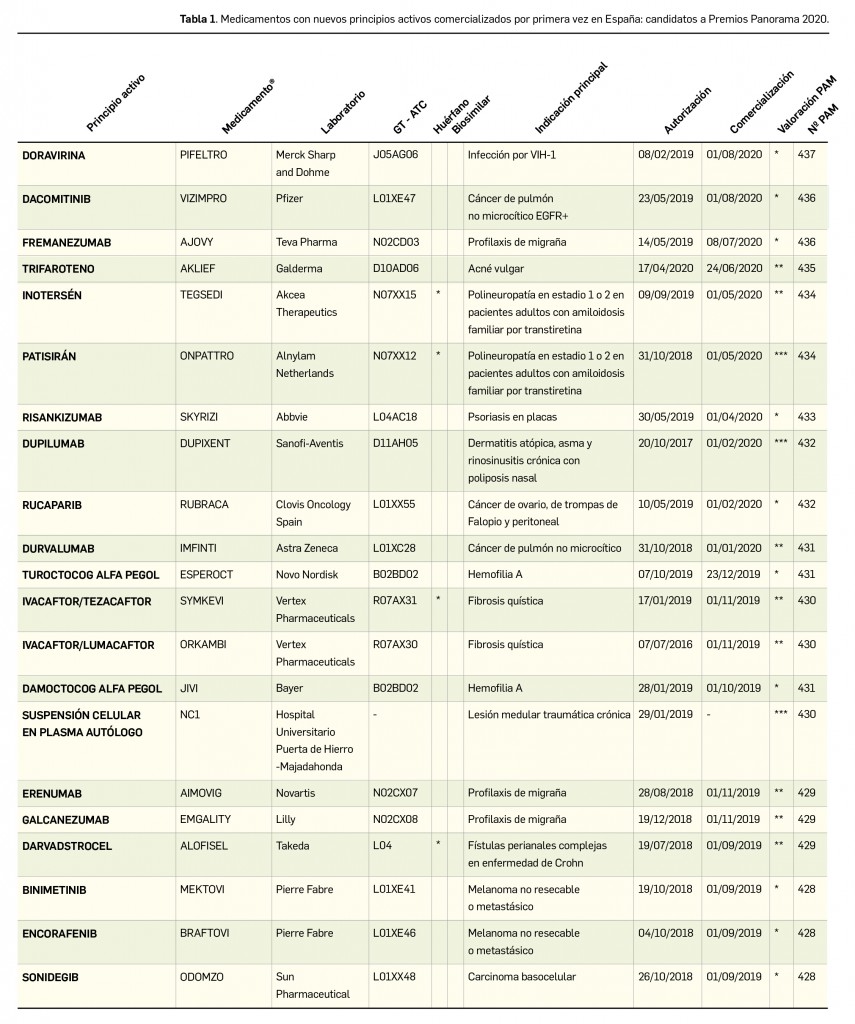
El periodo analizado transcurre entre el 1 de septiembre de 2019 y el 1 de septiembre de 2020, a partir de los datos recogidos en el Nomenclátor Oficial de la Prestación Farmacéutica del Sistema Nacional de Salud. En estos 12 meses, un total de 21 medicamentos con nuevos principios activos han sido comercializados por vez primera en España (Tabla 1), todos los cuales han sido revisados y evaluados anteriormente, y las correspondientes monografías fueron publicadas en Panorama Actual del Medicamento a lo largo del periodo mencionado.
Tras la discusión de los méritos de cada uno de los medicamentos estudiados, en la que participan todos los miembros del Jurado, éste acuerda lo siguiente:
– Atendiendo a los méritos específicos de cada uno de los medicamentos reseñados, se concede por unanimidad el Premio Panorama 2020 a:
Por representar el primer tratamiento específicamente dirigido a contrarrestar la inflamación tipo 2, mediada por células Th2, inaugurando una vía terapéutica. Se trata de un anticuerpo monoclonal que se une a la subunidad α del receptor de la IL-4, impide la señalización mediada por la unión de esa citocina a su receptor tipo I (IL-4Rα/γc) y también por IL-4 e IL-13 a través del receptor tipo II (IL4Rα/IL-13Rα), e induce prometedores efectos antiinflamatorios. Tras demostrar una significativa superioridad clínica frente a placebo, ha sido autorizado, en pacientes de ≥ 12 años, para el tratamiento de la dermatitis atópica moderada-grave y como tratamiento de mantenimiento adicional para el asma grave con inflamación de tipo 2 no adecuadamente controlada; patologías de alta prevalencia e impacto socio-económico y curso crónico-recurrente que afectan a población pediátrica y adulta y que, en casos graves, pueden resultar incapacitantes. En dermatitis atópica representa el primer avance en varios años (el primer tratamiento biológico autorizado), emergiendo como una opción interesante de tratamiento sistémico de novo en pacientes refractarios a medicación tópica o en aquellos pre-tratados en los que el uso de ciclosporina no es adecuado. En las dos indicaciones, se beneficiarán del fármaco, a priori, un elevado número de pacientes con patología severa en los que no se dispone de más opciones terapéuticas.
– Adicionalmente, se concede una Mención de Honor a:
Por ser el primer ARN pequeño de interferencia autorizado para el tratamiento de una enfermedad en humanos, cabeza de serie de una vasta clase de fármacos con gran potencial terapéutico en di-versas enfermedades, desde la hipercolesterolemia a algunos tipos de cáncer, que supondrán un im-portante avance en la medicina de precisión en los próximos años. Designado como medicamento huérfano, ha sido autorizado para el tratamiento de la polineuropatía en estadios 1 o 2 en pacientes adultos con amiloidosis familiar (o hereditaria) por transtiretina, una enfermedad rara en la que solo se disponía de una opción farmacológica –tafamidis– y en la que será una prometedora alternativa en los casi dos tercios de pacientes no candidatos a trasplante hepático (que se mantiene como el estándar de tratamiento en muchos casos); en comparación con tafamidis, amplia, además, el es-pectro de pacientes que pueden beneficiarse, por incluir también a aquellos con polineuropatía en estadio 2. Actúa mediante su unión específica a una secuencia conservada genéticamente en la re-gión 3′ no traducida del ARNm de la proteína transtiretina (TTR), en su forma mutante y salvaje, y a través de la interferencia de ARN prduce la degradación catalítica de dicho ARNm: determina la inhi-bición de la síntesis hepática y su secreción a sangre, lo que se traduce en una reducción sustancial de sus niveles séricos circulantes y una mayor estabilización o aclaramiento de los depósitos de TTR amiloidótica y de las manifestaciones de la polineuropatía.
Las monografías con la evaluación de estos nuevos medicamentos han sido publicadas previamente en los números 432 (Dupixent®) y 434 (Onpattro®) de Panorama Actual del Medicamento. Ambos han sido considerados como medicamentos que suponen una innovación importante (***), suponiendo una aportación sustancial a la terapéutica estándar. A modo de recordatorio y resumen, se recogen a continuación los principales aspectos innovadores de ambos.
Dupilumab es un nuevo anticuerpo monoclonal humano de administración por vía subcutánea que se une específicamente a la subunidad α del receptor de la interleucina 4 (IL-4Rα). Impide así la señalización molecular mediada por la unión de esa citocina a su receptor tipo I (IL-4Rα/γc) y también la señalización de IL-4 e IL-13 a través del receptor tipo II (IL-4Rα/IL-13Rα). Puesto que IL-4 e IL-13 son los principales mediadores de la inflamación tipo 2 –regulada por linfocitos Th2–, dupilumab ejerce efectos antiinflamatorios bloqueando esta ruta de señalización, que juega un papel fundamental en patologías relacionadas con la atopia. En base a ello, el medicamento ha sido autorizado para el tratamiento de la dermatitis atópica (DA) moderada-grave en pacientes adultos y adolescentes de ≥12 años candidatos a tratamiento sistémico, y también como tratamiento de mantenimiento adicional para el asma grave con inflamación de tipo 2 (caracterizada por eosinófilos elevados en sangre y/o FeNO elevado), en adultos y adolescentes de ≥ 12 años que no están adecuadamente controlados con corticosteroides inhalados en dosis altas en combinación con otro medicamento.
Los datos conducentes a su autorización en DA derivan de tres amplios estudios pivotales de fase 3, doblemente ciegos y controlados por placebo, que incluyeron pacientes adultos con enfermedad moderada-grave y refractarios a tratamiento tópico; la población estudiada puede considerarse representativa de la población diana a la cual se dirige el fármaco. Dos de esos estudios evaluaron el tratamiento en monoterapia y demostraron la superioridad de dupilumab frente a placebo tras 4 meses de tratamiento, con diferencias significativas en la proporción de pacientes que mejoran clínicamente: un aumento del 27-28% de respondedores según la escala IGA y del 37-44% en la proporción de los que alcanzan EASI-50 (aumentos del 23-28% para EASI-90). Un tercer estudio probó que dupilumab también es significativamente superior a placebo en combinación con un corticoide o inhibidor de calcineurina tópicos, con una eficacia que se mantiene por periodos de tratamiento de hasta 1 año (aumento de respondedores del 24-28% según escala IGA y de 40-48% en el número de pacientes con EASI-50 y de 28-36% con EASI-90).
Los resultados de un estudio controlado en adolescentes (12-17 años) corroboran la eficacia de dupilumab en este grupo etario, con aumentos frente a placebo del 22% y el 28% en la proporción de pacientes respondedores según escala IGA y que alcanzan EASI-75, respectivamente. En general, los efectos en el aclaramiento de la piel con el fármaco se acompañaron de mejorías notables del prurito (del orden del 25-30%), así como de la calidad de vida y los síntomas referidos por el paciente. El beneficio que aporta el fármaco, que parece instaurarse de forma rápida (2 semanas) y se mantiene en el tiempo, se verificó en los distintos subgrupos analizados, independientemente de factores como edad, sexo, raza o tratamiento de base; de forma interesante, los datos clínicos ponen de manifiesto la consistencia de la eficacia en pacientes con respuesta inadecuada, intolerancia a ciclosporina o en los que dicho fármaco no es aconsejable: tras 4 meses de tratamiento, alcanzaron EASI-75 un 59-69% de los pacientes vs. 18-29% con placebo.
Así pues, dupilumab ha demostrado superioridad frente a placebo en el tratamiento de la DA tanto en pacientes que inician tratamiento sistémico de novo tras fracaso del tratamiento tópico como en pacientes pretratados con ciclosporina, el fármaco que la mayoría de guías clínicas establecen de elección para el inicio del tratamiento sistémico (por la amplia experiencia de uso) y el único con eficacia contrastada y autorización para ello en la UE. No obstante, no se dispone de comparación directa de dupilumab con este fármaco (de hecho, la principal limitación de los estudios comentados es la ausencia de comparador activo) y los datos no permiten establecer la eficacia relativa entre ellos, por lo cual, hasta no se demuestre superioridad clínica, parece que ciclosporina1 se mantiene como el tratamiento de elección a corto plazo –máximo de 1 año– en la DA moderada-grave; a largo plazo los datos son más limitados y controvertidos, por el riesgo de nefrotoxicidad irreversible.
Con respecto a su uso en asma grave no controlada como tratamiento adicional (a corticoides inhalados más otro fármaco), la aprobación por la EMA se basó en los resultados de dos estudios controlados y doble ciego de fase 3. Un amplio ensayo de grupos paralelos demostró la superioridad de dupilumab frente a placebo en tratamientos de 1 año de duración, con una notable reducción de la tasa de exacerbaciones graves y mejora de la funcionalidad pulmonar, que en la práctica real se puede traducir en menor incidencia de hospitalizaciones y visitas a urgencias por crisis agudas. Esa eficacia, de inicio precoz (2 semanas) y duradera, se confirmó en el conjunto de pacientes incluidos, pero fue más pronunciada en la población con niveles basales elevados de biomarcadores inflamatorios de tipo 2 (altos niveles de eosinófilos en sangre y de FeNO en el aire exhalado). Así, en pacientes con ≥ 300 eosinófilos/µl, dupilumab redujo el riesgo de exacerbaciones en un 66-67% (tasa de 0,37-0,40 vs. 1,08-1,24 eventos/año con placebo) y duplicó el valor de VEF1 pre-broncodilatador (0,43-0,47 l vs. 0,21-0,22 l con placebo). Valores similares se observaban en pacientes con FeNO ≥ 50 ppb: tasa de 0,33-0,39 exacerbaciones/año (vs. 1,06-1,27 con placebo) y VEF1 pre-broncodilatador de 0,30-0,39 l (vs. 0,19-0,23 l con placebo).
El segundo estudio reveló que en el 70% de los pacientes tratados con dupilumab el asma mejora hasta el punto de que pueden reducir la dosis de corticoides orales (CSO), frente al 42% de los tratados con placebo; de hecho, la mayoría de quienes recibieron el fármaco pudieron reducir a la mitad la dosis de CSO (80% vs. 50% con placebo) y casi la mitad pudieron suprimir por completo el uso de CSO (48% vs. 25% con placebo), manteniendo un buen control del asma. En esta indicación la eficacia del fármaco también fue consistente en los distintos subgrupos analizados, destacando un mayor beneficio en pacientes con ≥ 150 eosinófilos/µl y con ≥ 25 ppb de FeNO. De forma interesante, los resultados comunicados por los pacientes del control del asma y la calidad de vida en los dos estudios –a través de los cuestionarios ACQ-5 y AQLQ(S)– muestran un aumento notable de la proporción de pacientes respondedores a dupilumab en comparación con placebo.
Por tanto, el nuevo fármaco representará una alternativa terapéutica interesante a otros anticuerpos que se oponen a los efectos de la IL-5 (mepolizumab, reslizumab y benralizumab) o la IgE (omalizumab), autorizados para pacientes con alto recuento de eosinófilos o demostración de asma alérgica mediada por IgE, respectivamente, pero que no han demostrado eficacia en un amplio rango de pacientes asmáticos cuya enfermedad está mediada por una compleja red de inflamación celular. No se dispone de comparaciones directas e indirectas con estos fármacos, siendo difícil el posicionamiento de su eficacia relativa por el distinto perfil de los grupos de pacientes que se benefician clínicamente de ellos, a priori más amplio en el caso de dupilumab. En cualquier caso, su uso como tratamiento adicional se restringe a pacientes con mal control a pesar de dosis altas de CSI (más otro fármaco), pues según las guías actuales, en pacientes tratados con dosis medias se recomienda aumentar dicha dosis antes de iniciar fármacos biológicos; en ese sentido, no implica un cambio sustancial en la terapéutica estándar. No obstante, los CSI no son capaces de normalizar los niveles de biomarcadores de inflamación tipo 2, y un 30% de pacientes con asma grave requieren tratamiento adicional con corticoides orales: en ellos, dupilumab sí puede representar una ventaja sustancial, permitiendo reducir/suprimir su dosis y, con ello, su toxicidad.
En términos de seguridad, dupilumab presenta un perfil toxicológico bien definido, relativamente benigno a corto-medio plazo y clínicamente manejable, que determina unas tasas bajas de abandono del tratamiento (< 6%), y que es consistente entre las dos indicaciones y dosis autorizadas del fármaco, con independencia de la edad de los pacientes. En línea con la tolerabilidad de otros fármacos biológicos empleados en patologías con componente inflamatorio, destacan por su frecuencia reacciones adversas como reacciones en el lugar de la inyección (eritema, edema y prurito) e infecciones (conjuntivitis, blefaritis, nasofaringitis, infecciones respiratorias del tracto superior, sinusitis y herpes oral), que en su mayoría son leves-moderadas y autolimitadas. El riesgo de desarrollo de reacciones alérgicas e inmunogenicidad parece bajo (la aparición de anticuerpos antifármaco en menos del 10% de pacientes no se relacionó con problemas concretos de seguridad o eficacia), si bien los datos de uso a largo plazo del fármaco –periodos de tratamiento de >1 año– son limitados y se requieren futuros estudios para descartar riesgos potenciales de desarrollo de neoplasias malignas.
En definitiva, dupilumab inaugura una nueva vía farmacológica en el tratamiento de la dermatitis atópica y del asma (a la que previsiblemente se incorporarán otros prometedores fármacos ya en desarrollo clínico, como lebrikizumab), patologías de curso crónico-recurrente que afectan a población pediátrica y adulta y que en determinados casos pueden resultar incapacitantes y tener un elevado impacto socio-laboral. A la innovación mecanística se suma el beneficio clínico superior a placebo demostrado en casos graves-moderados, siendo quizás más relevante su indicación en dermatitis atópica, patología en la cual no se han incorporado avances terapéuticos desde hace mucho tiempo y representa el primer tratamiento biológico autorizado: emerge como una opción adecuada de tratamiento sistémico de novo en pacientes refractarios a medicación tópica o en aquellos pre-tratados pero en los que el uso de ciclosporina no se considere adecuado por falta de respuesta, contraindicación o intolerancia.
Patisirán es un ácido ribonucleico pequeño de interferencia bicatenario formulado en nanopartículas lipídicas que facilitan su estabilidad y llegada al hígado, donde se une específicamente a una secuencia conservada genéticamente en la región 3′-UTR no traducida del ARNm de la proteína TTR, también tanto en su forma mutante como salvaje, y a través de la interferencia de ARN (con mediación de la endonucleasa argonauta-2) produce la degradación catalítica de dicho ARNm. Por tanto, determina una inhibición de la síntesis hepática de la proteína TTR y de su secreción a sangre, lo que se traduce en una reducción sustancial de los niveles séricos circulantes de la proteína y, con ello, en una mayor estabilización o aclaramiento de los depósitos de TTR amiloidótica y de las manifestaciones de la polineuropatía y cardiomiopatía.
El efecto farmacológico de este fármaco es novedoso en el sentido de que reduce los niveles tanto de la TTR de tipo mutado como salvaje, a diferencia del trasplante hepático ortotópico, que solo reduce la síntesis y niveles circulantes de proteína mutante (el órgano implantado seguirá sintetizando proteína TTR salvaje). Designado por la EMA como medicamento huérfano, ha sido autorizado para el tratamiento –por vía intravenosa una vez cada 3 semanas– de la polineuropatía en estadios 1 o 2 en pacientes adultos con amiloidosis familiar (o hereditaria) por transtiretina (ATTRh).
La eficacia clínica de patisirán ha sido adecuadamente contrastada en un ensayo pivotal de fase 3 (APOLLO), también doble ciego y controlado con placebo (N= 225). Los pacientes adultos con polineuropatía en estadios 1 o 2 por ATTRh tratados con dicho fármaco durante año y medio mostraron una significativa mejoría o estabilización de las manifestaciones en comparación con placebo, revelada por una diferencia de 34 puntos en la escala mNIS+7 (cambio de -6 puntos vs. +28 puntos con placebo; p< 0,001) y apreciable ya desde los primeros 9 meses (diferencia de 16,0 puntos frente a placebo); al final del estudio, la proporción de pacientes respondedores según el umbral de mNIS+7 fue ampliamente mayor con patisirán (56% vs. 4%). Su superioridad sobre placebo se confirmó en los resultados de todas las variables secundarias, destacando su efecto, por ejemplo, sobre el cambio en la puntuación del cuestionario Norfolk QoL-DN (diferencia de -21 puntos a favor de patisirán), en la prueba de la marcha de 10 m o en el índice de masa corporal modificado; además, el análisis por subgrupos demostró que esa eficacia es independiente de factores como edad, sexo, raza, tipo de mutación TTR, tratamiento previo y estadio de la enfermedad.
En términos de seguridad, patisirán es un fármaco bien tolerado, con bajas tasas de interrupción (1,4% vs. 0% con placebo) o retirada (5% vs. 14%) del tratamiento, similares o inferiores a placebo. Aunque los eventos adversos se relacionaron con el tratamiento en la mitad de pacientes (49% vs. 39% con placebo), la gran mayoría fueron limitados en el tiempo y leves-moderados, con escasa incidencia de los graves (2,7% vs. 0%). Las reacciones adversas más frecuentes fueron edema periférico (30% vs. 22%) y reacciones relacionadas con la perfusión2 (19% vs. 9%), entre las que destacan: dolor de espalda, rubefacción, dolor abdominal o náuseas. Patisirán no afecta al recuento de plaquetas ni a la funcionalidad renal y hepática, y presenta una reducida inmunogenicidad (solo presentaron anticuerpos el 4% de los pacientes), que no altera su perfil beneficio-riesgo. Un estudio abierto de extensión, en desarrollo a día de hoy, aportará una mayor información sobre su seguridad a largo plazo.
El ensayo clínico mencionado tuvo un diseño (tamaño, comparador, variables, etc.) y desarrollo que se considera aceptable por la EMA en el contexto de una enfermedad rara como la ATTRh, que progresivamente conduce a la muerte, lo cual complica la replicación de resultados en un segundo estudio. A la vista de los hallazgos comentados, se considera que la eficacia de patisirán en las manifestaciones clínicas y la calidad de vida de los pacientes con polineuropatía es clínicamente relevante, no así para los pacientes que solo presenten cardiomiopatía. Cabe destacar que ha sido investigado solo en pacientes con polineuropatía en estadio 1 o 2 (que aún caminan), o sea, no ha demostrado eficacia en los pacientes más graves en estadio 3 (alteración sensorio-motora generalizada, completamente dependientes, postrados en cama o silla de ruedas); pero a diferencia de tafamidis, sí será útil para el tratamiento de pacientes en estadio 2 (afectación de extremidades inferiores y manos, pueden todavía moverse con ayuda).
Aunque está por ver la influencia definitiva sobre la evolución global de la enfermedad a largo plazo (no hay datos sobre el impacto en supervivencia) y sus resultados clínicos puedan ser modestos, no cabe duda de que patisirán aporta un mecanismo de acción novedoso e inaugura una nueva vía terapéutica en el manejo de la ATTRh, potencialmente útil en otros cuadros de amiloidosis asociados al depósito de fibrillas de proteínas anómalamente plegadas. Es, pues, un nuevo tratamiento etiológico modificador del curso de la enfermedad cuya incorporación al mercado en una indicación en la que solo se disponía de una opción farmacológica (tafamidis) –útil además en una población más reducida de pacientes– es ciertamente relevante, especialmente si se considera que hasta dos tercios de los pacientes no son susceptibles de trasplante hepático (que se mantiene como el estándar terapéutico en muchos casos).
No se dispone aún de comparaciones clínicas directas entre las 3 opciones aprobadas para el tratamiento de la polineuropatía por ATTRh (tafamidis, patisirán e inotersén), lo que suele complicarse en enfermedades raras por los tamaños de muestra y costes asociados. Tampoco se han realizado comparaciones indirectas o meta-análisis que faciliten el posicionamiento de las distintas opciones; algunos autores incluso advierten de la dificultad de que éstos aporten una robustez aceptable (siempre limitada) dada la heterogeneidad entre los estudios pivotales de los tres fármacos que, aunque similares en diseño, tienen diferencias importantes en duración del tratamiento, criterios de selección de pacientes, características basales de los mismos o evaluación de la eficacia (Samjoo et al., 2020).
Teniendo en cuenta, por tanto, que no se puede aún concluir sobre la superioridad de uno u otro fármaco y por ahora se presentan como alternativas de tratamiento con distinto perfil de seguridad y pauta posológica, los resultados divulgados por una reciente revisión de la Cochrane (Magrinelli et al., 2020) y los datos comentados en este artículo podrían sugerir que inotersén y patisirán son más eficaces que tafamidis y pueden emplearse en un abanico más amplio de pacientes; han mostrado eficacia incluso en pacientes pre-tratados con tafamidis, por lo que su uso como 1ª línea condicionaría la posibilidad de emplear tratamientos posteriores en caso de progresión.
Si se compararan, de forma indirecta y no ajustada, los resultados numéricos de los dos nuevos fármacos, inotersén y patisirán, este último parece aportar un mayor beneficio clínico en términos de estabilización de las manifestaciones de la neuropatía periférica y de la calidad de vida de los pacientes.
Por último, habiéndose aprobado previamente otros oligonucléotidos antisentido (con el mismo fundamento que inotersén) para el tratamiento de otras patologías, el hecho de que patisirán sea el primero de su clase –el primer ARN de interferencia que se autoriza para tratamiento en humanos– hace que represente una mayor innovación terapéutica.
El trastorno por déficit de atención e hiperactividad (TDAH) es el trastorno del neurodesarrollo más frecuente en la etapa infanto-juvenil: se estima que afecta en torno a un 7% de los niños en edad escolar, representando, por tanto, un motivo común de consulta en atención primaria y pediatría. Su etiopatogenia es multifactorial, y en ella se deben considerar factores genéticos, neuroquímicos y anatómicos, así como la influencia de factores ambientales. Desde el punto de vista clínico, los tres síntomas cardinales del TDAH son déficit atencional, hiperactividad e impulsividad, si bien es un trastorno muy heterogéneo que puede presentar distintas manifestaciones según edad, sexo, comorbilidades y características individuales del paciente.
En cuanto a su abordaje, no existe ninguna prueba gold-standard para el diagnóstico del TDAH, que debe basarse en una valoración clínica exhaustiva que incluya una anamnesis completa, exploración física y psicopatológica del paciente, y una evaluación de su entorno psicosocial. De hecho, el tratamiento, multimodal, se debe iniciar con modificaciones del entorno que disminuyan el impacto de los síntomas, reservando el uso de fármacos para cuando haya riesgo de deterioro en más de un ámbito de la vida del paciente. Para la intervención farmacológica, se dispone de fármacos estimulantes, como metilfenidato y lisdexanfetamina, y de fármacos no estimulantes, como atomoxetina y guanfacina. El presente artículo revisa en mayor profundidad todos los conceptos aquí resumidos.
El trastorno por déficit de atención e hiperactividad (TADH) es el trastorno neuropsicobiológico más frecuente de la infancia, apareciendo antes de los 12 años y presentando continuidad hasta la edad adulta. Este trastorno del neurodesarrollo afecta al individuo y a su entorno, interfiriendo en el progreso académico y social del individuo, pudiendo ocasionar una importante problemática social y psicológica durante toda la vida del paciente.
Desde el ámbito psicológico se aborda como una alteración de la función ejecutiva, término que engloba muchas capacidades gnósticas (atención, memoria, capacidad de planificación, etc.) necesarias para realizar tareas esenciales para el funcionamiento cotidiano, así como para atender y organizar los distintos pasos con el fin de lograr un objetivo, reflexionar sobre las posibles consecuencias antes de hacer algo o inhibir la respuesta inmediata y cambiarla por otra más apropiada.
La prevalencia del TDAH se estima que es de un 7,2% de niños en edad escolar, disminuyendo en la adolescencia, por la dificultad de discriminar ciertas conductas más impulsivas propias de esta etapa de la vida. Se considera que la relación entre sexo masculino y femenino es 2 a 1, probablemente, por un infradiagnóstico en niñas, ya que en ellas los problemas de conductas son más sutiles y larvados; en los últimos años se plantea que es posible que la prevalencia en ambos sexos sea similar, aunque predomine el “subtipo inatento” en mujeres.
El origen neurobiológico del TADH ha sido demostrado en múltiples estudios, caracterizado por la existencia de deficiencias anatomo-biológicas, que afectan a ciertas estructuras. No obstante, la etiología del TDAH puede ser de origen genético o adquirido, dando lugar a las mismas alteraciones bioquímicas como base del trastorno: se encuentran defectos en los alelos de los genes que codifican los receptores de dopamina y otros neurotransmisores en el sistema nervioso central. Se han hallado patrones de heredabilidad en estos defectos genéticos. Además, en una serie de niños adoptados se ha observado la presencia de una etiología combinada, por un lado, constitucional, y por otra, adquirida, con la probabilidad de heredar patologías comórbidas (violencia, conflictividad, adicciones, etc.).
En relación con factores de riesgo, existe una asociación entre el TDAH y la prematuridad, infecciones del sistema nervioso central y algunas cromosomopatías, como el síndrome de Williams Beuren y “síndrome X frágil” entre otros. Los factores ambientales contribuyen a matizar la expresión clínica del trastorno, y la influencia de las circunstancias sociales y familiares contribuyen a exacerbar o mitigar la sintomatología y a condicionar la evolución del trastorno.
En resumen, pese a que no se conoce la causa exacta y no existe prueba complementaria para establecer el diagnóstico definitivo, se han identificado distintos factores relacionados con su génesis, que se describen a continuación.
Se han postulado teorías neuropsicológicas que tratan de explicar la conducta del paciente con TDAH, entre las que destaca el modelo del déficit de las funciones ejecutivas, planteado por Barkley, que propone la existencia de dificultades en la inhibición de respuestas que debe dar el paciente ante una situación determinada. El modelo del déficit motivacional, por su parte, indica que los niños con TDAH precisan de gratificación inmediata. Por último, el modelo de Brown recalca las dificultades en la función ejecutiva, pero matizada por aspectos emocionales y motivacionales. Dichos modelos no son excluyentes unos de otros, de modo que pueden coexistir en los diferentes subtipos debido a la heterogeneidad del TDAH.
Los tres síntomas cardinales del TDAH son déficit atencional, hiperactividad e impulsividad. A pesar de todo, el TDAH es muy heterogéneo, presentando distintas manifestaciones clínicas según edad (Tabla 1), sexo, comorbilidades y características individuales del paciente. La incapacidad para mantener la atención es el síntoma que durante más tiempo acompaña al individuo a lo largo de su vida, mientras que la impulsividad e hiperactividad predominan más durante la niñez. Conviene recordar que los síntomas se aprecian fácilmente en situaciones en las que se necesita un esfuerzo mental o mantener la atención de manera continuada, mientras que disminuyen en momentos organizados, novedosos y estructurados. Es por ello que, cuando el alumno con TDAH goza de la supervisión individual adecuada, presenta una disminución de los síntomas y un mejor rendimiento académico.
La versión más reciente del Manual Diagnóstico y Estadístico de los Trastornos Mentales (Diagnostic and Statistical Manual of Mental Disorders, DSM), el DSM V, incluye cambios significativos con respecto a la versión DSM IV TR del año 2000. En base a ellos, para que un paciente sea diagnosticado con TDAH debe cumplir las características clínicas que se definen a continuación.
A. Patrón persistente de inatención y/o hiperactividad-impulsividad que interfiere con el funcionamiento o desarrollo que se caracteriza por inatención (1) y/o hiperactividad e impulsividad (2),
1. Inatención: se define por la presencia de 61 (o más) de los siguientes síntomas se han mantenido durante, al menos, 6 meses en un grado que no concuerda con el nivel de desarrollo y que afecta directamente las actividades sociales y académicas/laborales:
2. Hiperactividad e impulsividad: 6 (o más) de los siguientes síntomas mantenidos durante, al menos, 6 meses en un grado que no concuerda con el nivel de desarrollo del paciente y que afecta directamente las actividades sociales y académicas/laborales:
B. Algunos síntomas de inatención o hiperactivo-impulsivos estaban presentes antes de los 12 años.
C. Varios síntomas de inatención o hiperactivo-impulsivos están presentes en 2 o más contextos (por ejemplo, en casa, en el colegio o el trabajo, con los amigos o familiares, o en otras actividades).
D. Existen pruebas claras de que los síntomas interfieren con el funcionamiento social, académico o laboral o reducen la calidad de los mismos.
E. Los síntomas no se producen exclusivamente durante el curso de la esquizofrenia o de otro trastorno psicótico y no se explican mejor por otro trastorno mental (por ejemplo, trastorno del estado de ánimo, trastorno de ansiedad, trastorno disociativo, trastorno de la personalidad, intoxicación o abstinencia de sustancias).
Además, la nueva versión del DSM (DSM V) describe tres presentaciones del TDAH:
1) Predominantemente inatento: impera la lentitud en el procesamiento de la información. Más frecuente en mujeres, pero debido a la ausencia de trastornos conductuales se suele retrasar el diagnóstico. Esta forma suele ir asociada a trastornos del humor y ansiedad.
2) Predominantemente hiperactivo-impulsivo: más común en niños en la etapa pre-escolar, es el precursor del subtipo combinado.
3) Tipo combinado: es el más prevalente, en que el individuo presenta características de ambas presentaciones clínicas previamente citadas. Es también el que más se parece a las descripciones clásicas del trastorno.
Se debe subrayar que, en aproximadamente un 50% de los casos, el TDAH se acompaña de otros trastornos psiquiátricos comórbidos, principalmente trastorno negativista-desafiante, trastornos de ansiedad, depresivos y trastornos de aprendizaje. Además, los pacientes con TDAH suelen desarrollar con mayor frecuencia: baja autoestima, consumo de tabaco, alcohol y otros tóxicos, trastornos del sueño, peor adhesión a dietas saludables, riesgo de obesidad y sobrepeso, accidentes y menor esperanza de vida por causas naturales, accidentes y suicidio. Por lo general, las mujeres presentan con mayor frecuencia trastornos emocionales y menos problemas conductuales que los varones, motivo por el cual el diagnóstico se suele retrasar.
Tanto el diagnóstico precoz como el tratamiento adecuado influyen de forma positiva en los pacientes, ayudando a prevenir todas las complicaciones descritas. No obstante, cabe destacar que el hecho de padecer TDAH puede conllevar una serie de aspectos positivos en los niños/adolescentes, tales como su espontaneidad, simpatía, mayor energía o entusiasmo.
—El TDAH es el trastorno neuropsicobiológico más prevalente en la infancia—
El diagnóstico del TDAH es exclusivamente clínico, pues no existen pruebas complementarias con evidencias diagnósticas definitivas: las pruebas de imagen, neurofisiológicas o de laboratorio no están indicadas a menos que haya una sospecha clínica razonable. Por tanto, la valoración clínica ha de ser exhaustiva e incluir una anamnesis completa, exploración física y psicopatológica del paciente, así como una evaluación del entorno psicosocial del individuo.
La exploración física debe incluir peso, talla, IMC, pulso, tensión arterial, visión, audición, rasgos dismórficos, lesiones cutáneas y discromías. Podemos encontrar algunos signos que sugieren la presencia de determinados síndromes (X frágil, Williams, síndrome alcohólico-fetal). Conocer el estado basal del paciente es necesario para, posteriormente, valorar efectos secundarios si se inicia tratamiento farmacológico.
Nunca debe usarse una respuesta positiva a la medicación como herramienta diagnóstica. Resulta imprescindible recabar información de distintos ámbitos y de diferentes observadores (padres y profesores), ya que los síntomas pueden variar y oscilar según la actividad y el entorno. El empleo de escalas y cuestionarios auto-aplicados para padres y profesores ayudan en la evaluación del paciente, aunque no se debe realizar el diagnóstico teniendo en cuenta solamente la respuesta a las escalas o cuestionarios.
Se dispone de dos tipos de cuestionarios, unos específicos para TDAH y otras herramientas más generales con el fin de realizar una evaluación global del individuo. Entre los primeros, destacan el cuestionario EDAH (debe ser rellenado por el profesor del paciente) y el ADHD rating scale–IV (deben rellenarlos los padres).
Por otra parte, la evaluación neuropsicológica no puede usarse para confirmar o descartar el TDAH, sino que solo sirve para apoyar la evaluación clínica, arrojar información sobre la situación del paciente (capacidad cognitiva, lectoescritura, atención, planificación, flexibilidad cognitiva), realizar el diagnóstico diferencial y como ayuda en el soporte terapéutico. Entre los cuestionarios para dicha evaluación destacan el Continous Permormance Test, el Stroop colour–Word–interference Test, el de figura completa de Rey-Osterrieth, el test de Dígitos directos e inversos, el test de caras, etc. Además, una evaluación psicométrica mediante el coeficiente intelectual con el cuestionario WISC puede ser de gran ayuda para planificar los apoyos al alumno, ya que nos ofrecerá información acerca de la fortaleza y puntos débiles del paciente.
Los síntomas cardinales del TDAH (déficit de atención, hiperactividad e impulsividad) no son patognomónicos de este trastorno por lo que es necesario descartar otros procesos que se solapan con el TDAH, de ahí la importancia de la correcta realización de la evaluación clínica para el diagnóstico diferencial (Tabla 2).
Por otra parte, el pronóstico del TDAH será más favorable cuando predomine la inatención sobre la impulsividad e hiperactividad, no se desarrolle conducta antisocial y las relaciones con familiares y otros compañeros sean adecuadas. En la Tabla 3 se exponen los factores de riesgo para desarrollar TDAH y los factores que los padres debieran promover durante la infancia del paciente para hacer frente a los de riesgo, a fin de mejorar el pronóstico.
El tratamiento de elección en el TDAH es multimodal e incluye la intervención psicosocial y la intervención farmacológica.
La intervención psicosocial comprende un conjunto de programas que han demostrado su efectividad en la resolución de problemas que acompañan al TDAH. Incluye todas aquellas actividades que permiten una mejoría en el rendimiento atencional y en el rendimiento escolar y que ayudan a manejar la conducta del paciente. El apoyo escolar, mediante medidas de refuerzo, adaptaciones del método, y estrategias para mejorar el rendimiento y la motivación, ha demostrado ser una herramienta de suma importancia en el manejo de estos pacientes.
Por su parte, el tratamiento farmacológico se debe considerar en todo paciente con TDAH en que el diagnóstico esté bien establecido, en base a los criterios clínicos actuales y a una repercusión clínica, ya sea en su rendimiento escolar, relación social, autoestima o en la calidad de vida. En España, disponemos de tratamientos psicoestimulantes y no estimulantes. Entre las novedades, dentro del primer grupo, destaca la lisdexanfetamina, y en el segundo grupo, la guanfacina de liberación retardada.
Los fármacos estimulantes son seguros y eficaces en el tratamiento del TDAH. Se administran vía oral, se absorben a nivel gastrointestinal y son lipofílicos, por lo que cruzan la barrera hematoencefálica. En cuanto a su farmacodinamia y mecanismo de acción, todos comparten el bloqueo del transportador y, con ello, de la recaptación sináptica de dopamina y noradrenalina, elevando sus niveles en el encéfalo; también aumentan la liberación de ambos neurotransmisores en el espacio intersináptico. Así, se ha probado que la anfetamina inhibe la recaptación, estimula la liberación, impide el almacenamiento en las vesículas de dopamina y noradrenalina, y revierte la dirección de acción del transportador, sacando activamente dopamina y noradrenalina a la sinapsis.
En definitiva, los estimulantes provocan una elevación de dopamina en el núcleo estriado, que media efectos motores, elevan la dopamina en el núcleo accumbens, donde media los efectos de recompensa, y elevan también dopamina en el córtex prefrontal, donde media los efectos terapéuticos beneficiosos sobre atención y memoria. Tres cuartas partes de los pacientes TDAH tratados con metilfenidato responden favorablemente, y, además de mejorar los rasgos característicos de la enfermedad, también implementan la función social, cognitiva y disminuyen la agresividad.
Entre ellos, por ejemplo, el metilfenidato de liberación inmediata alcanza el pico plasmático en 1-2 h y tiene una vida media de 3-6 h, más corta que la dextroanfetamina. Así, su efecto farmacológico se inicia entre 30 y 60 minutos, presentando el pico terapéutico entre 1 y 2 h, y desapareciendo a las 2-6 h; la duración máxima del efecto terapéutico es de 4 a 6 h. El metilfenidato se metaboliza por desesterificación hepática al metabolito ácido ritalínico y se elimina principalmente por la orina. La dosis eficaz en niños suele estar entre 1 y 2 mg/kg de peso/día, aunque se debe ajustar individualmente. Se suele iniciar con dosis de 18 mg/día y se recomiendan incrementos de 9-18 mg semanales hasta ajustar la dosis según la tolerabilidad, respuesta y peso del paciente. Las formulaciones de acción prolongada pueden evitar el efecto rebote y no desencadenan la sensación de tristeza al no tener un pico plasmático tan elevado.
Metilfenidato se presenta en:
Por otra parte, la lisdexanfetamina (Elvanse®, autorizada en España desde 2014) se reserva para niños mayores de 6 años que no han respondido bien a metilfenidato o que han presentado efectos secundarios. Se trata de un profármaco de dextroanfetamina que es inactivo fuera del cuerpo humano: tras su administración, se absorbe y, por acción de las enzimas eritrocitarias, se separa la dexanfetamina de la lisina, activándose el fármaco. Presenta una duración en su efecto farmacológico de unas 13 h, alcanzando su concentración máxima entre 1 y 3,5 h.
Lisdexanfetamina está disponible en cápsulas de 30, 50 y 70 mg, que permiten su apertura y dilución en agua para niños que no pueden tragarlas. Se recomienda empezar el tratamiento con la dosis de 30 mg e ir subiendo gradualmente según necesidad y tolerancia; no se debe tomar como referencia la dosis de otros fármacos para calcular la dosis inicial del tratamiento. La dosis eficaz de lisdexanfetamina en niños más pequeños suele ser de 30 mg al día, en niños mayores de 50 mg al día y en adolescentes de 70 mg diarios. Entre sus ventajas principales, destaca menor riesgo de abuso (perfil más seguro) y, entre los inconvenientes, el alto coste (requiere visado).
En breve podremos asistir a la comercialización de nuevas presentaciones en solución oral, jarabe o comprimidos masticables de liberación modificada para administración nocturna de metilfenidato y lisdexanfetamina.
Los efectos secundarios más frecuentes de los fármacos estimulantes son: insomnio de conciliación, pérdida de apetito, cefaleas y nerviosismo. Con respecto a la seguridad cardiaca, ningún estudio ha demostrado mayor incidencia de eventos cardiovasculares (incluyendo muerte súbita) en el grupo de pacientes con TDAH frente a grupo control. Por tanto, de entrada, no es necesario realizar electrocardiograma antes del inicio de tratamiento con estimulantes. No obstante, en niños con cardiopatía o arritmias conocidas debemos preguntar por la existencia de sintomatología cardiaca de reciente aparición tras el inicio de la medicación, en cuyo caso se debe derivar a cardiología para realizar ecocardiograma y prueba de estrés, ya que un electrocardiograma en reposo no descarta el problema.
Por otro lado, los fármacos no estimulantes se reservan para casos con riesgos de abuso del fármaco o en caso de no respuesta con estimulantes. Entre ellos, la atomoxetina se puede usar en niños y adolescentes a partir de los 6 años a dosis de 1,2-1,8 mg/kg/día en una sola toma. Los síntomas nucleares mejoran en menor medida que con los estimulantes y está indicada en pacientes con tics, trastorno de ansiedad o consumo de drogas. Alcanza el efecto tras 3 o 4 semanas. Los efectos adversos son leves e incluyen síntomas digestivos, irritabilidad y fatiga.
Por su parte, la guanfacina es un agonista α2A adrenérgico que parece mejorar la función del córtex prefrontal dorsolateral (que regula la atención y la acción) y de la corteza prefrontal ventromedial (que regula la emoción). La guanfacina de acción modificada (Intuniv®, autorizado en 2015), que se presenta en comprimidos de 1, 2, 3 y 4 mg, mejora la sintomatología del paciente con solo una dosis diaria, favoreciendo el cumplimiento frente a la guanfacina clásica. La mayor eficacia se ha documentado con dosis de 0,1 mg/kg/día, aunque dosis menores podrían ser igual de eficaces. Se recomienda evitar incrementos superiores a 1 mg/semana, siendo la dosis máxima de 7 mg/día. El riesgo de abuso es prácticamente inexistente. Es obligado que sea tragada, aunque se puede administrar indistintamente en el desayuno, merienda o cena. Se precisa vigilar la aparición de efectos secundarios, entre los que destaca la hipotensión arterial, sintomatología digestiva, cefalea y fatiga. La guanfacina ha demostrado, además, ser muy útil en el control de la sintomatología del trastorno negativista desafiante y en el control de los tics.
El cáncer infantil es un término genérico que designa a un amplio grupo de enfermedades neoplásicas –que, a priori, pueden ser de todo tipo– desarrolladas en la edad pediátrica (< 18 años). Se trata de una de las principales causas de mortalidad entre niños y adolescentes de todo el mundo; concretamente, la primera en niños de 0 a 14 años de edad. Cada año se diagnostican hasta 300.000 nuevos casos en pacientes de entre 0 y 18 años en todo el mundo, más de 1.100 en España. Entre ellos, los tumores hematológicos (que afectan a casi la mitad de los niños menores de 15 años que padecen cáncer) y los del sistema nervioso central son los más prevalentes, no asociándose en una amplia mayoría a causas o factores de riesgo concretos, pues solo un 10% se relacionan con una predisposición genética. En conjunto, se trata de problemas de salud que, además de la relevancia sanitaria y económica, conllevan un elevado impacto psicológico y emocional para pacientes, familiares y para toda la sociedad.
Pero, afortunadamente, su mortalidad es relativamente baja: se estiman tasas de supervivencia a los 5 años cercanas al 80% en los países de nuestro entorno, y que alcanzan el 100% en algunos tipos concretos de tumores. Se ha demostrado que el diagnóstico precoz es vital en la mejora del pronóstico, a fin de asegurar un acceso temprano al tratamiento especializado en unidades hospitalarias y multidisciplinares de onco-hematología pediátrica; ello requiere de la concienciación de familias y profesionales sanitarios, de un acceso equitativo a la atención sanitaria y de una mejora en la evaluación clínica y estadificación de los tumores. Si bien en los últimos años se han incorporado avances importantes en el manejo de algunos tumores infantiles, destacando la inmunoterapia (y, en particular, los medicamentos CAR-T), la mayoría de ellos se siguen tratando con las terapias oncológicas “clásicas”, como cirugía, radioterapia y quimioterapia.
Por su interés, el presente artículo aborda una revisión de la epidemiología del cáncer pediátrico en España y revisa los principales aspectos fisiopatológicos y clínicos de las neoplasias más frecuentes en niños y adolescentes: los cánceres hematológicos. Centra el foco fundamentalmente en la terapéutica farmacológica y los últimos avances incorporados en el manejo de las neoplasias hematológicas más frecuentes en este grupo etario: leucemia linfoblástica aguda, leucemia mieloide aguda, linfoma de Hodgkin y linfomas no-Hodgkin. Se revisa también el papel asistencial que el profesional farmacéutico puede desarrollar para con los pacientes y sus familiares, en términos de educación sanitaria, detección precoz y optimización de la farmacoterapia.
La oncología pediátrica engloba un grupo variado de enfermedades neoplásicas, tanto hematológicas como tumores sólidos, que afectan a niños y adolescentes, cada una con su propio diagnóstico, pronóstico y tratamiento, pero caracterizadas todas ellas por proliferación descontrolada de al menos un tipo celular. De forma general, se habla de “niños” entre las edades de 0 y 14 años y de “adolescentes” entre los 15 y los 18 años. En todos los casos, son problemas de salud que, además de su elevado impacto sanitario y económico, conllevan una alta carga psicológica y emocional para los familiares de los pacientes y para toda la sociedad, y a los que no siempre se les presta la atención que merecen por quedar ocultos en la abundancia de los cánceres propios de la edad adulta.
Hablando, en líneas generales, de “cáncer pediátrico”, se trata de una patología considerada poco frecuente en esa población, aunque en los últimos años viene presentando un aumento considerable. Según datos de la UICC (Unión Internacional Contra el Cáncer), actualmente se diagnostican más de 250.000 nuevos casos de niños y adolescentes con cáncer en el mundo –pueden representar entre el 1% y el 3% de todos los casos de cáncer–, perteneciendo unos 15.000 diagnósticos al continente europeo; no obstante, algunos autores apuntan a que hasta el 80% de ellos se dan en países en vías de desarrollo. Se ha llegado a considerar la primera causa de mortalidad en los niños entre los 5 y 14 años a nivel mundial, también en los países desarrollados, causante de más de 90.000 muertes anuales (más de 3.000 en Europa). A principios de este siglo, los fallecimientos por cáncer pediátrico constituían ya el 5% de años potenciales de vida perdidos para el conjunto de pacientes con cáncer entre los 0-70 años, siendo solamente superado en este aspecto por el cáncer de mama.
Se desconoce con exactitud la incidencia y evolución del cáncer pediátrico en España, y en mayor medida en población adolescente, pero el Registro Español de Tumores Infantiles (RETI-SEHOP)1ha supuesto un gran avance en su estudio epidemiológico. Aunque no recoge la totalidad de los casos, se estima que refleja actualmente una cobertura de más del 90% de los tumores infantiles en España, aproximándose al 100% de los casos en algunas regiones (Aragón, Cataluña, País Vasco, Madrid y Navarra).
Su último informe, presentado en 2018 y con datos relativos al periodo comprendido entre 1980 y 2017 (puede tener limitaciones debidas a una cobertura menor 70% en las últimas décadas del siglo XX), refleja que las enfermedades neoplásicas infantiles más prevalentes (Figura 1) son las leucemias, siendo la más común la leucemia linfoblástica aguda, seguido de los tumores sólidos del sistema nervioso central y de los linfomas, considerándose el linfoma No Hodgkin el más frecuente en la edad pediátrica (Pardo et al., 2018). En ese periodo se han notificado un total de 30.715 casos en pacientes de 0 a 19 años, por lo que se estima que en los últimos años se notifican en torno a 1.200-1.400 nuevos casos de cáncer en dicho grupo etario. Esto se traduce en una tasa de incidencia bruta para todos los tumores de 157 casos nuevos al año por cada millón de niños de 0-14 años (tasa estandarizada de 159,4 casos/millón), una cifra de incidencia muy similar a la de otros países cercanos de la UE, como Francia y Alemania, si bien representan solo entre el 0,5 y el 1% del total de neoplasias.
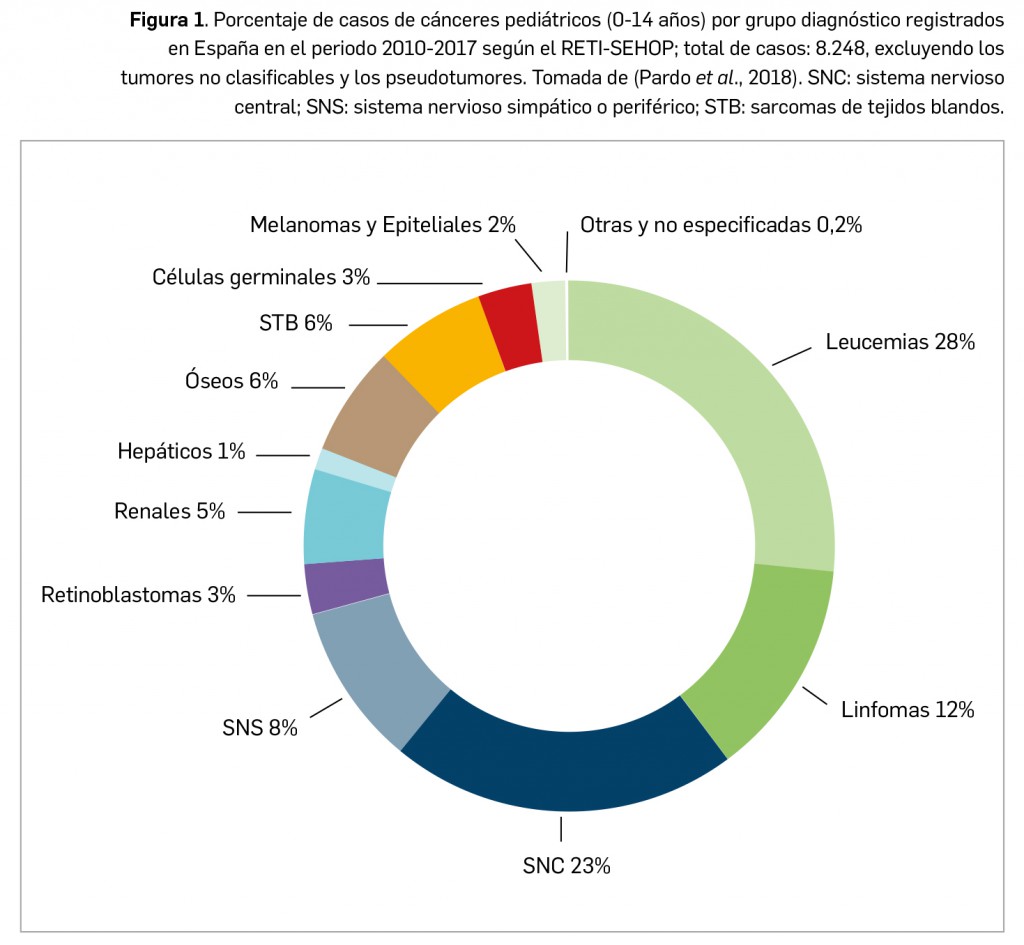
En concreto, según el citado Registro, los cánceres más incidentes en niños pre-adolescentes (0-14 años) son, en este orden, la leucemia linfoblástica aguda, los astrocitomas, los neuroblastomas, la leucemia mieloide aguda, los nefroblastomas y los linfomas Hodgkin y no Hodgkin (Pardo et al., 2018). En la etapa adolescente (15-19 años), sin embargo, se ha descrito una incidencia variable, sobre todo en lo referente a tumores sólidos (Figura 2).
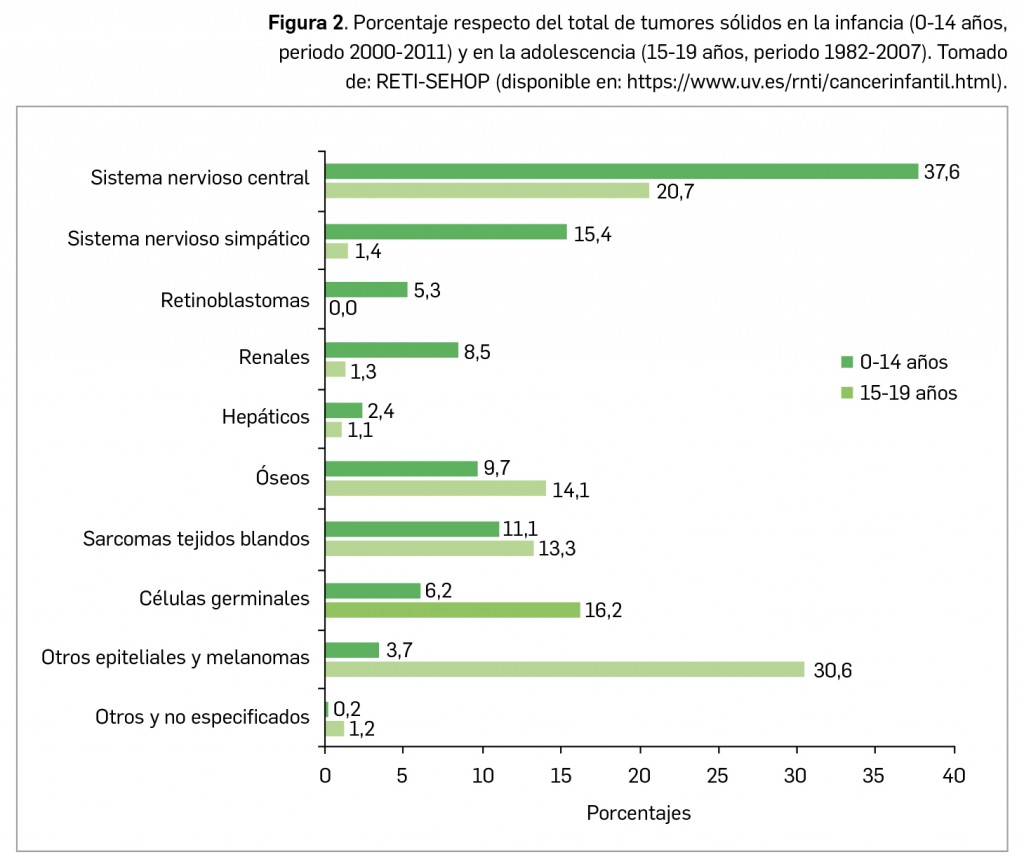
Aunque pueden afectar por igual a niños que a niñas, en la incidencia de tumores infantiles en España hay un predominio del sexo masculino (57% de los casos). En cuanto a la edad de diagnóstico, la mayoría de los casos notificados en el periodo evaluado (29.227) se refieren a niños de menos de 14 años; concretamente, casi la mitad de las neoplasias se presentan antes de los 4 años de edad: el 11% de los casos se dan en el primer año de vida, el 36% entre 1-4 años, el 28% entre 5-9 años y el 25% entre 10-14 años. A este respecto, una de las principales características de los tumores en población pediátrica es que, si se compara con la edad adulta (en que los tumores suelen desarrollarse más lentamente, con periodos de latencia de hasta varias décadas en algunos casos), la proliferación celular es más rápida y agresiva debido al carácter embrionario e inmaduro de las células tumorales (Ferrís et al., 2004).
En cuanto a su etiología, los escasos factores de riesgo confirmados explican muy pocos casos de cáncer infantil. Como en la mayoría de tumores diagnosticados en edad adulta, el origen se considera multifactorial, incluyendo una variedad de factores medioambientales (físicos, químicos y biológicos) y genéticos, cuya interacción es todavía ampliamente desconocida. Se ha postulado que algunas alteraciones genéticas predisponentes –identificadas en cerca de un 10% de los niños con cáncer– podrían tener origen medioambiental y algunos factores (exposición a radiaciones ionizantes, sustancias químicas como benceno o tabaco, y virus) podrían actuar antes del nacimiento o de la concepción. Sin embargo, hasta hoy los estudios epidemiológicos no han podido identificar factores de riesgo medioambiental específicos (dificultados por la caracterización y cuantificación de las exposiciones preconcepcionales, concepcionales, transplacentarias y posnatales), que, aunque se creen asociados a casi el 90% de los cánceres infantiles, aún necesitan confirmación.
Afortunadamente, uno de los aspectos más positivos en el abordaje de los cánceres infantiles es su pronóstico. Grosso modo, a día de hoy se consideran curables un 80% de los casos (ascendiendo hasta un 90-100% para algunos tipos concretos de cáncer, como retinoblastomas o linfomas de Hodgkin), gracias a los avances incorporados en técnicas diagnósticas, en el tratamiento y en el cuidado integral de los pacientes oncológicos pediátricos en las últimas cuatro décadas; también contribuye el hecho de que los niños suelen tolerar mejor los tratamientos que los pacientes adultos. Así, la tasa global de supervivencia a 5 años en niños con cáncer (0-14 años) en España alcanza casi el 80% para aquellos diagnosticados en 2005-2006, estimándose una mejora de casi 23 puntos porcentuales desde 1980, lo que ha supuesto una reducción a la mitad del riesgo de mortalidad (Figura 3). En la cohorte de pacientes diagnosticados en 2009-2011, esa supervivencia a 5 años se estimó en el 79%, ascendiendo al 84% en la supervivencia a los 3 años.
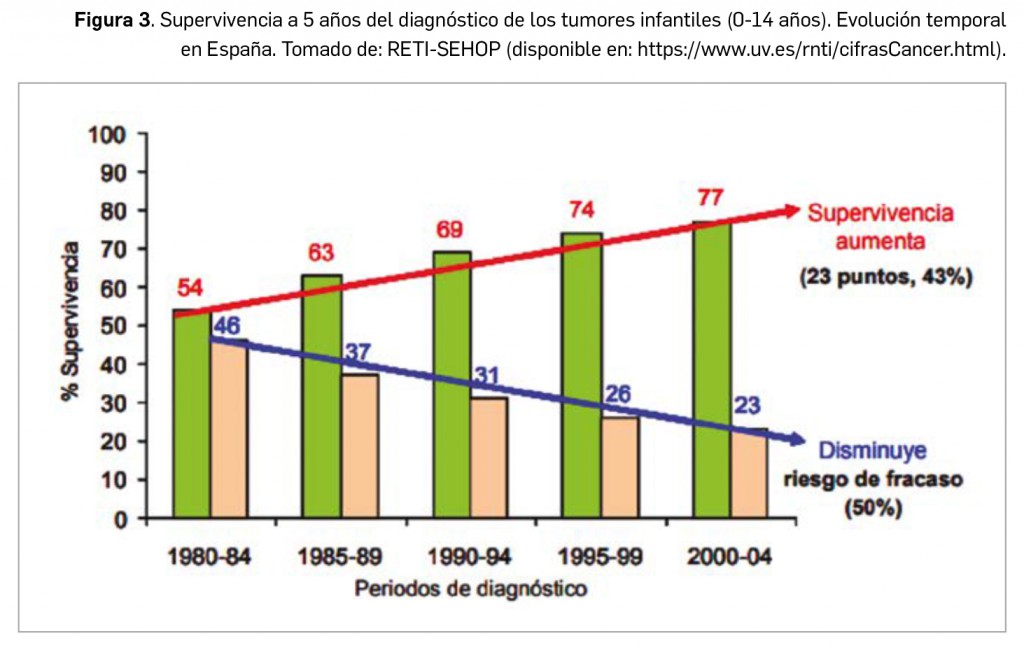
De forma general, en nuestro entorno el pronóstico del cáncer infantil se ha estabilizado en las últimas 2 décadas, si bien se han reportado algunas diferencias notables en las tasas de supervivencia a 5 años variables entre países (por ejemplo, 44,9% en Letonia vs. 90,1% en Islandia para la cohorte de casos diagnosticados en 1990-94) y regiones europeas (por ejemplo, 62% en el este vs. 77% en el norte para la cohorte 1988-97). En contraste, las tasas de curación en los países subdesarrollados, en los que se concentran una mayoría de diagnósticos (≈80%), son de tan solo el 10-20%, esto es, solo 1-2 niños de cada 10 sobrevivirán. Entre las principales causas de tal diferencia en estos países pueden identificarse: el acceso restringido al centro de tratamiento, una menor concienciación social y de los profesionales sanitarios, el coste de los medicamentos y las limitaciones de cuidados oncológicos especializados.
Se estima que unos 500.000 ciudadanos europeos son supervivientes de un cáncer pediátrico, pudiendo alcanzar fácilmente el millón hacia 2025. Hay que tener en cuenta que muchos de estos pacientes reciben tratamientos relativamente “antiguos”, con fármacos descubiertos hace más de 30 años, algunos de los cuales implican riesgos a largo plazo, de forma que un 20-40% de pacientes sufre algún tipo de secuela (Pardo et al., 2018). Por ejemplo, algunos autores advierten de la necesidad de que todos los niños tratados de un cáncer deban ser monitorizados durante la pubertad por el riesgo de que aparezcan hallazgos clínicos y analíticos sugestivos de insuficiencia gonadal, habida cuenta de que no hay un marcador clínico, hormonal o ecográfico suficiente para determinar si existe insuficiencia gonadal, lo que obliga a que el seguimiento de estos niños se prolongue a la edad adulta (Ibáñez et al., 2007).
Por otra parte, ante la imposibilidad actual de una prevención primaria o secundaria del cáncer infantil, la lucha contra estas enfermedades reside en la tarea diagnóstica y terapéutica. Lo más común es que la sospecha de un cáncer en el niño derive de la consulta de pediatría, abordándose posteriormente el diagnóstico mediante pruebas analíticas, estudios de imagen (radiografía, ecografía, resonancia magnética o tomografía axial computarizada) e incluso gammagrafías; la mayoría de casos requerirán del análisis de las células tumorales por medio de una biopsia confirmatoria del diagnóstico.
Tal y como se ha sugerido previamente, cabe destacar que muchos hospitales de referencia en nuestro país y los de nuestro entorno tienen unidades especiales de tratamiento de oncohematología pediátrica formadas por equipos multidisciplinares (incluyen desde oncólogos infantiles, radiólogos, cirujanos o intensivistas, hasta farmacéuticos, enfermeros, psicólogos y trabajadores sociales) para el abordaje especializado de estos pacientes. No obstante, muchos adolescentes diagnosticados entre 15 y 18 años son tratados en unidades de adultos a pesar de las recomendaciones de las sociedades profesionales y del Ministerio de Sanidad, que coinciden en que la atención de los pacientes oncológicos adolescentes debe realizarse de forma óptima en unidades pediátricas: la evidencia apunta a un mejor pronóstico cuando son tratados en ese entorno.
Además de las especificidades de los tratamientos –fundamentalmente son de tres tipos: cirugía, radioterapia y quimioterapia2– que se comentarán a continuación, el abordaje de los cánceres sanguíneos, principalmente las leucemias, requiere de un tratamiento de soporte de los pacientes, basado en el manejo y la prevención de las com-plicaciones surgidas durante el tratamiento específico. Incluye: la transfusión de componentes sanguíneos según las necesidades, medidas nutricionales individualizadas, el empleo de sistemas de aislamiento y antibioterapia profiláctica/terapéutica para la prevención/tratamiento de las infecciones durante el periodo de aplasia, la prevención del síndrome de lisis tumoral mediante la hidratación y el uso de agentes hipouricemiantes, y el tratamiento de la hiperleucocitosis mediante citorreducción con hidroxiurea o leucoaféresis. Puesto que el diagnóstico de cáncer infantil se asume como una enfermedad grave, no es infrecuente la incorporación de los cuidados paliativos desde el principio, con el objetivo de garantizar la máxima calidad de vida en el niño.
Por su relevancia epidemiológica, el presente artículo se centrará en los aspectos fisiopatológicos, clínicos y, sobre todo, de tratamiento de los tumores de mayor incidencia en la edad pediátrica: los cánceres hematológicos. Se estima que casi la mitad de los niños menores de 15 años que padecen cáncer sufren una leucemia o un linfoma, mientras que el otro 50% de los casos engloba diversos tumores sólidos diferentes (como los tumores cerebrales, osteosarcomas, tumor de Wilms, sarcoma de Ewing, rabdomiosarcoma o retinoblastoma), quedando éstos últimos fuera del enfoque de la presente revisión.
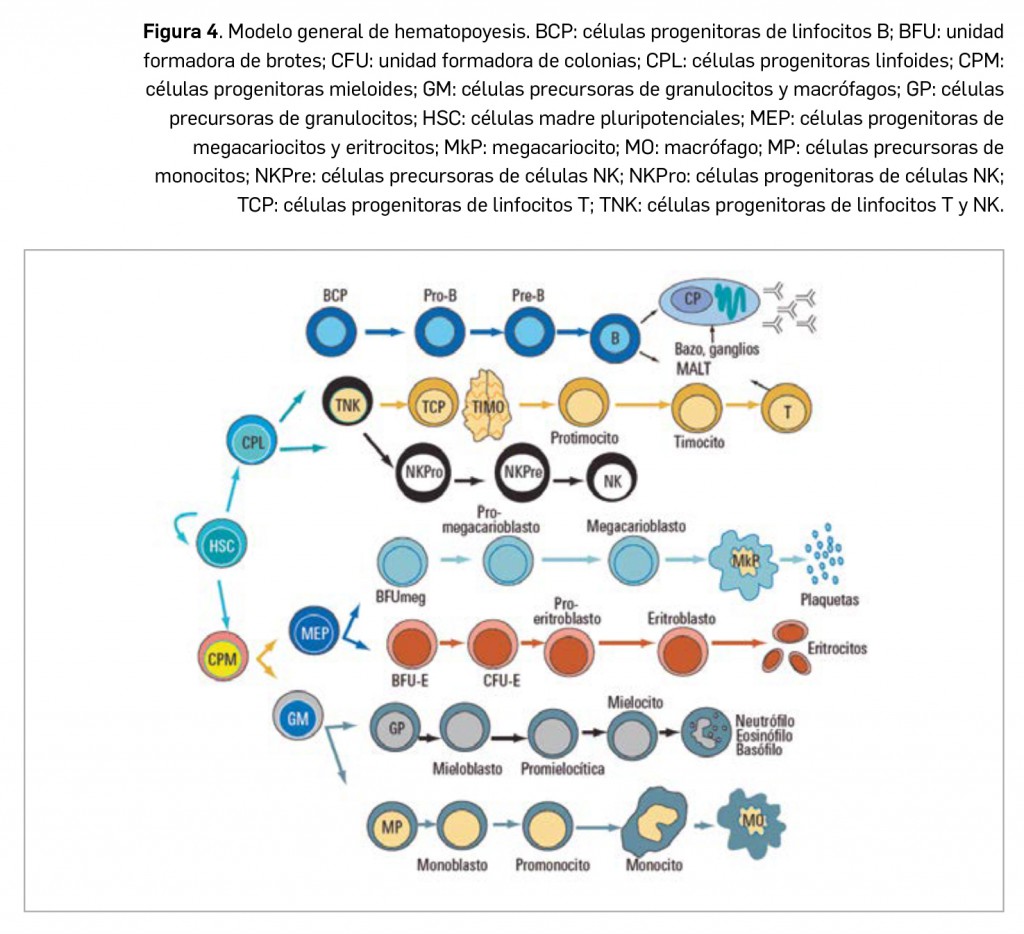
Bajo este término se engloban todos aquellos procesos de origen tumoral que afectan al tejido hematopoyético y al sistema linfoide. En general, se considera tejido hematopoyético a la médula ósea y todo su complejo sistema celular. Respecto al sistema linfoide, integra a los ganglios, tejido linfoide de diferentes órganos y bazo fundamentalmente, incluyendo sobre todo a los procesos que afectan a elementos celulares, como son los linfocitos B y T, y a las células plasmáticas (Figura 4).
De forma muy general, se podrían clasificar las neoplasias hematológicas siguiendo el esquema propuesto en la Tabla 1. Para una mayor precisión y especificidad en la clasificación del cáncer infantil, se recomienda consultar la nueva versión de Clasificación Internacional para Cáncer Infantil ICCC-3, disponible en la página web del Instituto Nacional del Cáncer estadounidense (https://seer.cancer.gov/iccc/iccc3.html), la cual ha incorporado, además de los tumores hematológicos (leucemias y linfomas), otros numerosos tipos de tumores.
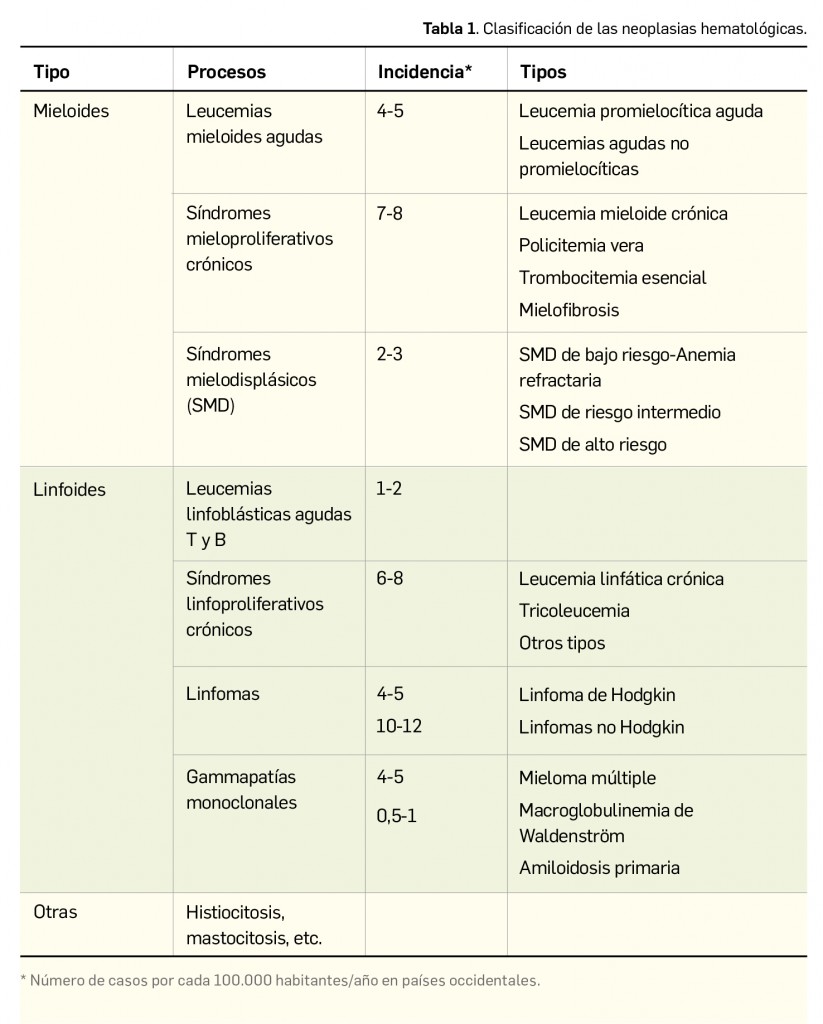
Como grupo, suponen algo más del 10% de los tumores en humanos, siendo más frecuentes en general en la edad avanzada (la incidencia se incrementa notablemente en casi todos los casos, multiplicándose unas 10 veces a partir de los 80 años, sobre todo en leucemias agudas y gammapatías), aunque, como se verá, hay excepciones. No obstante, a diferencia de tumores sólidos, no se dispone de registros exhaustivos de algunos de estos procesos, posiblemente porque acontecen en edad muy avanzada o porque se trata de procesos neoplásicos poco agresivos que conviven con otras enfermedades de base del paciente y no son comunicados (por ejemplo, leucemia linfática crónica en los estadios iniciales o los síndromes mielodisplásicos3).
A grandes rasgos, e independientemente de la edad de aparición, la causa de los tumores hematológicos es multifactorial y no se conocen con exactitud causas directas. Predominan eventos oncogenéticos primarios o secundarios que originan una pro-liferación descontrolada de un clon celular neoplásico. No existe un mecanismo genético molecular común, y solo en algunos procesos se conoce con exactitud el evento mutacional que da lugar a la enfermedad, lo cual es relevante para el abordaje terapéutico. Así, por ejemplo, en el linfoma de Burkitt se puede apreciar una translocación entre alguno de los cromosomas de los pares 8 y 14 –t(8;14)– que se traduce en la activación de un protooncogén (c-MYC); en el linfoma folicular puede detectarse una t(14;18) que sobreexpresa Bcl2, responsable del bloqueo de apoptosis de las células tumorales; o en el linfoma de células del manto, puede aparecer una t(11;14), con sobreexpresión de Bcl-1 (ciclina D1). Además, como en todas las neoplasias, se han implicado algunos factores ambientales, como las radiaciones ionizantes, sustancias químicas, como benceno o pesticidas, que pueden influir en su aparición. Lo que sí está claro es que el tratamiento previo con quimio/radioterapia por otra neoplasia o las situaciones de inmunodeficiencia conllevan un mayor riesgo de padecerlas (Alegre et al., 2017).
La leucemia linfoblástica aguda o leucemia agua linfoblástica (LAL o LAL) se produce por una proliferación incontrolada de un clon celular inmaduro dentro de la linfopoyesis (linfoblastos), que infiltra la médula ósea e invade la sangre periférica y otros órganos. Se divide en varios subtipos, cuya clasificación fue actualizada en 2016 por la Organización Mundial de la Salud (OMS) según se refleja en la Tabla 2.

Se trata, con diferencia, del grupo de neoplasias más frecuentes en niños (representa casi un tercio de los casos), con una incidencia aproximada de 4 casos nuevos/100.000 habitantes/año en los países desarrollados. Concretamente, en España en el periodo 2000-2016 se ha recogido una incidencia anual de 3,6 casos por cada 100.000 habitantes de entre 0 y 14 años. Además, se ha observado que la incidencia en niños es mayor entre los 3 y los 5 años, mientras que en adultos tiene una incidencia de 3/100.000/año, predominando en adultos jóvenes (25-30 años) y de sexo masculino.
Al igual que ocurre con el resto de las leucemias agudas, los síntomas se establecen de manera aguda, en un plazo de no más de 3 meses antes del diagnóstico, y la clínica deriva de la infiltración de los distintos órganos por las células leucémicas. En esta entidad es más frecuente respecto a otras leucemias la presencia de adenopatías (inflamación de los ganglios linfáticos), hepatomegalia y esplenomegalia (aumento de tamaño del hígado y del bazo, respectivamente). En cualquier caso, comparte con otras enfermedades leucémicas agudas las manifestaciones debidas a las citopenias por insuficiencia medular, esto es, anemia (que se manifiesta con palidez, astenia, taquicardia, disnea, etc), neutropenia (predisposición a infecciones, fiebre) y trombopenia (hemorragias a distintos niveles). La infiltración de órganos extramedulares también puede provocar cefalea o meningismo (si infiltra el sistema nervioso central), sarcomas granulocíticos (piel), ceguera (ojos) o leucostasis (pulmones).
Tal y como ocurre con las leucemias mieloides agudas, existe un sustrato molecular y citogenético que produce la evolución descontrolada del clon maligno de células y las consiguientes manifestaciones, siendo la etiopatogenia en ambas patologías superponible, con el resultado de acúmulo de blastos en la médula ósea. Estas células son capaces de multiplicarse y proliferar, pero han perdido la capacidad de diferenciarse a células hematopoyéticas maduras. La hipótesis más aceptada para explicar el desarrollo de la leucemia es la de “doble hit”: por un lado, existe una mutación que confiere capacidad proliferativa y, por otro lado, una mutación que altera la diferenciación hematopoyética. Además, mutaciones en los genes reguladores epigenéticos también juegan un papel fundamental en la leucemogénesis.
Las anomalías cromosómicas conducentes a la leucemia suelen consistir básicamente en translocaciones e inversiones de trozos de cromosomas que provocan la formación de nuevo material genético susceptible de generar nuevas proteínas de fusión, las cuales pueden perder su función original o ejercerla de forma incontrolada, escapando a los sistemas de regulación de la expresión génica. En el caso de que estas proteínas anómalas actúen como factores de transcripción, se produce una interrupción de la diferenciación y la posterior proliferación incontrolada.
Los principales factores pronósticos de la LAL son: la edad, precisamente más favorable en niños y adultos jóvenes; el recuento de leucocitos, con peor pronóstico en caso de hiperleucocitosis; el fenotipo, siendo desfavorables aquellas de fenotipo T y la pro-B; y la citogenética, ya que las hiperploidías tienen mejor pronóstico y las hipoploidías y algunas alteraciones genéticas, peor. La rápida respuesta al tratamiento confiere también un me-jor pronóstico, así como lograr una disminución rápida y mantenida de la enfermedad mínima residual.
El tratamiento de la LAL logra, especialmente en niños, unos porcentajes de remisión completa –definida morfológicamente como presencia de < 5% de blastos en el aspirado de médula ósea y recuperación de los recuentos celulares en el hemograma– superiores al 90%, y permite alcanzar tasas de pacientes libres de enfermedad, y probablemente curados, a los 5 años superiores a un 70%. En concreto, el informe RETI-SEHOP ha estimado una supervivencia a 5 años del 83% para aquellos pacientes diagnosticados en España en la cohorte de 2009-2011.
De forma general, para éste y otros tipos de leucemia aguda se sigue un tratamiento secuencial. La obtención de la remisión completa marca una inflexión crucial en la evolución, el tratamiento y el pronóstico, pero en la mayoría de los casos oculta una población celular leucémica residual tan importante como para producir inexorablemente una recaída si no se administra más tratamiento. Por ello, el tratamiento posremisión consiste en una serie de fases consecutivas. En primer lugar, la consolidación, como se denomina al ciclo de quimioterapia que se administra inmediatamente después de la inducción tras alcanzar la remisión completa. En segundo lugar, la intensificación se refiere a la quimioterapia que se suele administrar después de consolidar la remisión, con dosis escaladas, con la intención de erradicar la enfermedad mínima residual que persista tras la consolidación. En tercer lugar, en ocasiones puede establecerse un tratamiento de mantenimiento, que es una forma de quimioterapia prolongada a dosis bajas, que pretende impedir el re-crecimiento e incluso erradicar lentamente la EMR.
El trasplante de progenitores –células madre– hematopoyéticos (TPH) autólogo o alogénico puede también emplearse como tratamiento final tras la intensificación.
No obstante, la LAL es una enfermedad heterogénea con diferentes subgrupos que muestran una respuesta variable a la quimioterapia y, aunque a continuación se expone la estructura genérica del tratamiento (Figura 5), a día de hoy la estrategia terapéutica se individualiza según los factores pronósticos, fundamentalmente la edad (infantil o de adultos), el subtipo inmunológico y la genética. La LAL es sensible a varios fármacos, por lo que se usan diversas combinaciones de los mismos y, a diferencia de la leucemia mieloblástica, se ha demostrado la utilidad del tratamiento de mantenimiento. Es obligatorio el tratamiento profiláctico de los santuarios, en particular del sistema nervioso central (SNC).
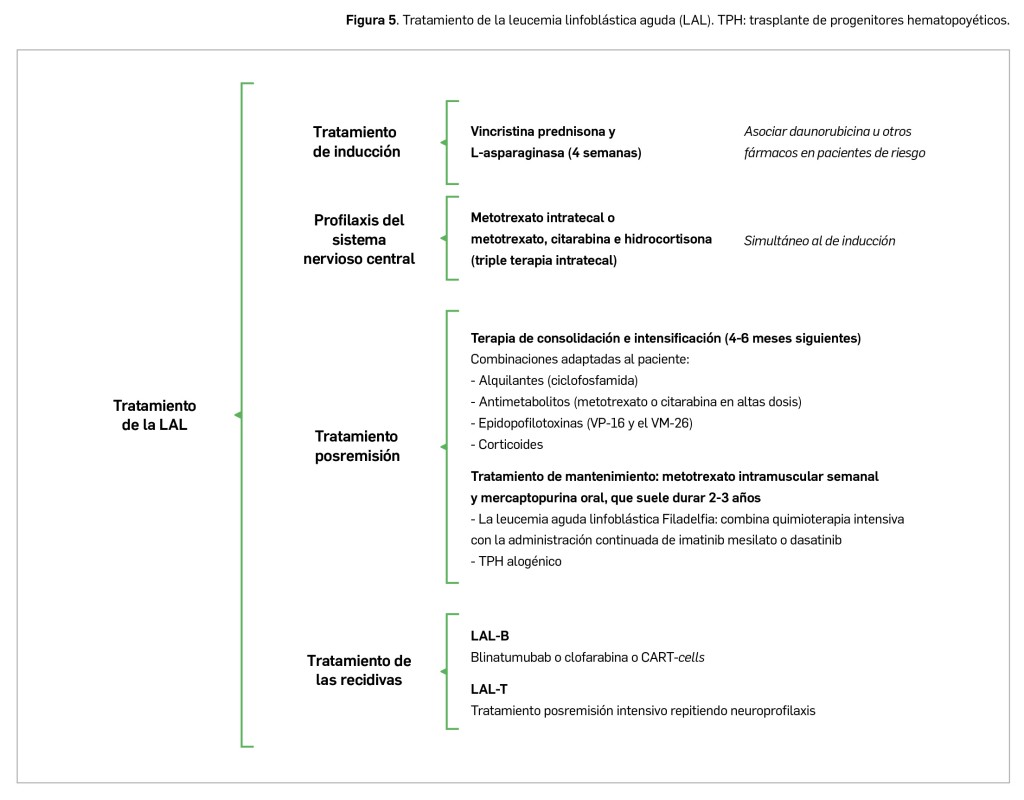
La combinación básica es la asociación de vincristina (EFG), prednisona (Dacortin® y EFG) y L-asparaginasa (Oncaspar®), que se administra a lo largo de 4 semanas. En los grupos de alto riesgo se asocia daunorubicina y otros fármacos. Con este esquema, más del 90% de los pacientes entran rápidamente en respuesta completa, siendo la lentitud en la respuesta o la persistencia de enfermedad mínima residual detectable por inmunofenotipo o citogenética uno de los factores pronósticos adversos más relevantes.
La meningitis leucémica es la forma de recaída de hasta el 60% de los niños con LAL si no reciben profilaxis del SNC. La quimioterapia sistémica atraviesa mal la barrera hematoencefálica, por lo que constituye un santuario donde los blastos leucémicos permanecen intactos, se reproducen localmente y, eventualmente, generan una recaída generalizada. La profilaxis del SNC se debe efectuar de forma rutinaria en esta entidad y consiste en inyecciones intratecales seriadas de metotrexato o, en algunos protocolos más intensivos, con una combinación de metotrexato, citarabina e hidrocortisona (triple terapia intratecal), que comienza ya durante la inducción. En este contexto, se han empezado a utilizar con buenos resultados formas liposomales de citarabina. En cambio, la irradiación craneal se ha abandonado como forma de profilaxis, debido a sus efectos adversos a largo plazo en el desarrollo intelectual y en el aprendizaje de los niños.
Una vez alcanzada la respuesta completa, se recomienda continuar con terapia de consolidación e intensificación durante los 4-6 meses siguientes. En la LAL existen multitud de protocolos distintos que combinan, en diversas formas y dosis, los fármacos eficaces para adaptarlos al riesgo diferencial de cada situación; entre ellos se encuentran: agentes alquilantes como la ciclofosfamida, antimetabolitos como el metotrexato o la citarabina en altas dosis, epidopodofilotoxinas como el VP-16 y el VM-26, y corticoides.
Acabada esta fase más intensiva, se pasa a un tratamiento de mantenimiento con metotrexato intramuscular semanal (Emthexate®, Metotrexato Accord®, Metotrexato Wyeth® y EFG) y mercaptopurina oral (Mercaptopurina Aspen® y Mercaptopurina Silver®), que suele durar 2-3 años. En los niños de riesgo estándar se pueden conseguir curaciones del 80% con una inducción y una consolidación no muy intensivas, con unos 2 años de mantenimiento suave. Por el contrario, los protocolos para los casos de mayor riesgo intensifican en gran medida el tratamiento de los primeros meses, aumentando el número de fármacos y sus dosis, tanto en la inducción como en las fases de consolidación e intensificación, y se siguen de un mantenimiento que periódicamente se intensifica con algún ciclo de altas dosis de quimioterapia combinada. Los resultados son siempre peores en adultos que en niños, incluso con factores pronósticos similares.
Un caso especial lo constituye la LAL Filadelfia4 positiva, que exige protocolos específicos en los que se combina quimioterapia intensiva –generalmente citarabina, altas dosis de CVDD (combinación de ciclofosfamida, vincristina, doxorrubicina y dexametasona) o FCG (fludarabina, citarabina y factor estimulante de colonias de granulocitos)– con la administración continuada de inhibidores de tirosina cinasa específicos como imatinib mesilato (Glivec® y EFG) o dasatinib (Sprycel®); en pacientes con intolerancia o resistencia a dastinib y en quienes no esté clínicamente indicado el tratamiento subsiguiente con imatinib, se puede recurrir a ponatinib (Iclusig®). No obstante, en este subtipo de LAL el pronóstico con quimioterapia es muy pobre, con tasas de supervivencia prolongada inferiores al 20%, por lo que tanto en adultos como en niños con buen estado de salud general está indicado el TPH alogénico en primera remisión, tras la inducción y la consolidación.
A pesar de un tratamiento adecuado, la leucemia puede recidivar en la médula ósea o en localizaciones extramedulares en hasta un 15-20% de los pacientes (25-30% en grupos de alto riesgo). Cabe destacar que hasta el 80% de los pacientes con recaída medular logran una segunda respuesta completa con el mismo tratamiento de inducción. No obstante, los pacientes con LAL complicada, en remisión tras más de una línea de tratamiento, constituyen una población de pronóstico pobre en quienes las opciones terapéuticas son muy escasas y la supervivencia se acorta de forma muy marcada. Como se verá más adelante, es precisamente en ese subgrupo de pacientes en el que se ha alcanzado un mayor progreso terapéutico en los últimos años, fundamentalmente para el tratamiento de LAL de células B recidivante.
El tratamiento de la LAL en recaída suele consistir en quimioterapia de rescate, a ser posible con una combinación de fármacos diferente a la usada en un inicio, seguida de TPH alogénico, que es la única opción potencialmente curativa, aunque muestra resultados de supervivencia global no excesivamente prometedores (del 20-45% a los 5 años). Además, en los casos de recaída tras trasplante (o pacientes no candidatos al mismo por edad, comorbilidades, falta de donante, refractariedad) no existe ningún tratamiento estándar además de la clofarabina –con resultados muy pobres– y el tratamiento paliativo. En el caso de la LAL de células T en recidiva hay aún menos opciones.
En cualquier caso, el tratamiento posremisión debe ser intensivo y es recomendable repetir la neuroprofilaxis para evitar la recaída extramedular en forma de leucemia meníngea. En los varones es también habitual la recidiva testicular, por lo que algunos protocolos incluyen la realización de biopsia testicular al final de la terapia de mantenimiento; el tratamiento de elección es la irradiación local. Tras la recaída ex-tramedular, existe un alto riesgo de recidiva generalizada, lo que hace imprescindible la repetición completa del tratamiento sistémico. En algunos pacientes se emplean tratamientos dirigidos, como los que se mencionan a continuación.
La European Medicines Agency (EMA) aprobó a finales de 2015 el uso de blinatumomab (Blincyto®), un anticuerpo monoclonal biespecífico diseñado para unirse selectivamente a CD19, que se expresa sobre la superficie de células de linaje B, y a CD3, que se expresa sobre la superficie de células T; así, activa células T endógenas conectando el CD3 del complejo del receptor de células T (TCR) con el CD19 de las células B benignas y malignas. La actividad antitumoral de blinatumomab no depende de que las células T sean portadoras de un TCR específico ni de los antígenos peptídicos que presentan las células cancerosas, sino que es de naturaleza policlonal e independiente del antígeno leucocitario humano (HLA) de las células diana: el fármaco actúa como mediador en la formación de una sinapsis citolítica entre las células T y las células tumorales, liberando enzimas proteolíticas para destruir tanto a las células diana proliferantes como en reposo.
Aunque aún no ha sido comercializado de forma efectiva en España, la administración por vía intravenosa de blinatumomab se asocia con una mayor producción transitoria de moléculas de adhesión celular, la producción de proteínas citolíticas, la liberación de citocinas inflamatorias y la proliferación de células T, teniendo como resultado la eliminación de las células CD19+. En base a ello, y a los resultados prometedores en pacientes en recidiva, ha sido indicado en monoterapia en pacientes adultos con LAL de precursores B con cromosoma Filadelfia-, CD19+ y en situación refractaria o en recaída, o bien en primera o segunda remisión completa. En pacientes pediátricos de > 1 año, se ha autorizado para el tratamiento en monoterapia de LAL de precursores B con cromosoma Filadelfia-, CD19+ y en recaída/refractariedad tras ≥ 2 tratamientos anteriores o un TPH alogénico. En estos pacientes, ha demostrado aportar una tasa de respuesta completa del 29%, que se traduce en una mediana de la supervivencia sin recaídas de 6,8 meses y de supervivencia global de 7,5 meses (AEMPS, 2018).
También se han autorizado otros agentes como la clofarabina (Evoltra® y EFG), un profármaco5 de antimetabolito nucleósido purínico aprobado para los casos de LAL en pacientes pediátricos de hasta ≤ 21 años tras el diagnóstico inicial, que estén en recidiva o refractarios tras ≥ 2 regímenes de tratamiento previos y para los que no existe ninguna otra opción terapéutica con la que se prevea una respuesta duradera. Se cree que su actividad antitumoral se debe a 3 mecanismos: a) inhibición de la ADN polimerasa α que da lugar a una terminación de la elongación de la cadena de ADN y/o de la síntesis/reparación del ADN; b) inhibición de la ribonucleótido reductasa, con la consiguiente disminución de los depósitos celulares de desoxinucleótido trifosfato (dNTP); y/o c) ruptura de la integridad de la membrana mitocondrial, con liberación de citocromo C y de otros factores proapoptóticos que llevan a la muerte programada de la célula, incluso de los linfocitos no proliferativos.
En ensayos clínicos de fase 2, la administración de clofarabina dio lugar a una reducción drástica y rápida de las células leucémicas periféricas en el 94% de los pacientes. La mediana de la supervivencia global de los 12 pacientes que alcanzaron una remisión completa (con o sin recuperación total de plaquetas) fue de 66,6 semanas, superando incluso las 100 semanas en quienes recibieron un trasplante mientras continuaban en remisión, aunque en ellos existen factores de confusión a la hora de valorar la duración de las respuestas. Se observó respuesta en distintos inmunofenotipos de LAL, incluidos los de células pre-B y los de células T (AEMPS, 2016).
Otra innovación incorporada recientemente que merece una reseña –aunque aún no se ha establecido su seguridad y eficacia en población pediátrica– ha sido inotuzumab ozogamicina (Besponsa®). Se trata de un nuevo fármaco conjugado compuesto por un anticuerpo monoclonal anti-CD22 humano, inotuzumab, que está enlazado covalentemente con la molécula pequeña N-acetil-γ-calicheamicina, un producto natural semisintético citotóxico (Figura 6). Así, se dirige específicamente contra las células que expresan CD22 en su superficie, incluyendo las células cancerosas pro B y pre B, y tras su internalización, libera el derivado de calicheamicina, cuya activación induce roturas del ADN bicatenario de la célula cancerígena, la interrupción del ciclo y la muerte celular por apoptosis. El medicamento, designado como huérfano, fue oficialmente autorizado para el tratamiento de adultos con LAL de precursores de linfocitos B CD22+ recidivante o refractaria; aquellos pacientes con cromosoma Filadelfia+ deben tener fracaso terapéutico con ≥ 1 inhibidor de la tirosina-cinasa (imatinib, dasatinib o ponatinib).
La eficacia del fármaco en la indicación y dosis autorizadas fue adecuadamente contrastada en un amplio ensayo pivotal de fase 3 (N= 326), multicéntrico y multinacional, aleatorizado, controlado y abierto. La administración por vía intravenosa de inotuzumab ozogamicina en monoterapia ha demostrado superioridad clínica frente a los regímenes de quimioterapia “clásica” seleccionados por el investigador, en términos de tasa de respuesta completa (81% en el brazo experimental vs. 29% en el brazo control) y, en menor medida, también de supervivencia global (mediana de 7,7 meses vs. 6,5 meses, que se traduce en un 23% menos de riesgo de muerte). Las variables secundarias confirmaron el beneficio del nuevo fármaco, destacando el aumento significativo del número de pacientes con negatividad para enfermedad mínima residual (78% vs. 28%) y de pacientes que pudieron ser sometidos a un trasplante de progenitores hematopoyéticos (48% vs. 22%), los cuales mostraron mejores cifras de supervivencia. Los resultados de remisión hematológica fueron consistentes entre los distintos subgrupos analizados, con independencia de la edad, número de tratamientos previos, duración de la remisión inicial, o refractariedad/recaída. Por otra parte, el perfil toxicológico del fármaco es importante, pero parece adecuadamente definido y se considera aceptable y clínicamente manejable, con una frecuencia de eventos adversos graves relacionados con el tratamiento similar a los de la quimioterapia control (31% vs. 29%). No obstante, un mayor número de pacientes necesitó interrupción (44% vs. 12%) o abandono del tratamiento (18% vs. 8%) en el brazo experimental, como consecuencia fundamentalmente de la neutropenia febril y los trastornos hepatobiliares venooclusivos. Estos últimos, junto a la toxicidad hepática grave, representan el mayor riesgo de seguridad asociado al nuevo fármaco.
En definitiva, inotuzumab ozogamicina inauguró una nueva vía mecanística en el tratamiento de la LAL de células B en una población diana (adultos en recaída o refractariedad) con un pobre pronóstico de supervivencia y escasas opciones terapéuticas. Sin representar una cura y con una modesta prolongación de la supervivencia global (1,5 meses), en comparación con la quimioterapia convencional incrementa significativamente la respuesta hematológica y las posibilidades de alcanzar el trasplante (única opción potencialmente curativa). Por tanto, se puede posicionar como alternativa de primera de elección (o segunda en pacientes refractarios a blinatumomab) en 2ª o 3ª líneas de tratamiento. Por el riesgo aparente de mortalidad precoz postrasplante, cuya relación con el fármaco aún no ha sido esclarecida, el Informe de Posicionamiento Terapéutico advierte de la necesidad de valorar muy cuidadosamente el beneficio-riesgo en cada paciente antes de utilizarlo (Fernández-Moriano, 2019a).
Pero, sin duda, el mayor progreso terapéutico en niños con LAL se ha dado en el campo de las terapias avanzadas. Así, en 2019 se comercializaba por primera vez en España tisagenlecleucel (Kymriah®), el primer medicamento de terapia génica a base de células CAR-T. Se trata de un novedoso tratamiento de inmunoterapia consistente en células T autólogas (del propio paciente) modificadas genéticamente ex vivo mediante el empleo de un vector lentiviral para expresar un receptor de antígeno quimérico (CAR) anti-CD19 (Revuelta et al., 2019) (Figura 7). Tras la unión de los linfocitos T reprogramados que expresan CAR a las células que expresan CD19 –células del linaje B desde etapas tempranas de su desarrollo a células plasmáticas, tanto malignas como normales–, la proteína quimérica transmite las señales intracelulares necesarias para activar la citotoxicidad frente a esas células CD19+6, así como una señal que favorece la expansión de las células T y la persistencia de tisagenlecleucel. El medicamento, designado como huérfano, ha sido oficialmente autorizado para el tratamiento, en perfusión intravenosa única, de pacientes pediátricos y adultos jóvenes de hasta 25 años de edad con LAL de células B refractaria, en recaída postrasplante o en segunda o posterior recaída, y de pacientes adultos con linfoma difuso de células grandes B (LBDCG) en recaída o refractario tras ≥ 2 líneas de tratamiento sistémico. Ambas indicaciones representan necesidades médicas no cubiertas.
La eficacia y seguridad clínicas de tisagenlecleucel en LAL fueron evaluadas mediante un ensayo pivotal de fase 2 (estudio B2202), de un solo brazo y abierto (sin comparador directo), con un número reducido de pacientes (N= 75). En dicha indicación de se disponía, además, de datos de otros dos ensayos clínicos fase 2 de soporte. A nivel general, tisagenlecleucel ha mostrado respuestas clínicamente relevantes en términos de tasa de respuesta global (69-95%, completa en el 60-75% de pacientes) como variable primaria de eficacia. Si bien los periodos de seguimiento de los pacientes han sido limitados y es difícil establecer la duración de la respuesta, se han obtenido datos de supervivencia global a 12 meses en el rango de 76-86% y medianas de supervivencia global de al menos 19 meses. Esto, unido a la escasa experiencia poscomercialización, impide aún considerarlo como un tratamiento curativo; los futuros resultados con un mayor seguimiento clarificarán si existe beneficio en términos de supervivencia libre de enfermedad a largo plazo, como así apuntan los indicios actualmente disponibles.
En cuanto a la seguridad clínica, el perfil toxicológico de tisagenlecleucel (influido por la quimioterapia previa a la perfusión) es importante, con un alto porcentaje de pacientes –cercano al 90%– que sufren reacciones adversas específicas graves (grado ≥ 3) o potencialmente mortales en el periodo posperfusión. El síndrome de liberación de citoquinas, las infecciones graves y los efectos adversos neurológicos son quizá los aspectos cruciales del manejo de su toxicidad, que debe ser abordado por profesionales sanitarios especialmente capacitados para el uso de estas terapias.
En cualquier caso, por su fundamento (los medicamentos CAR-T han supuesto un cambio de paradigma en el concepto de medicamento, que aquí son las propias células –modificadas– del paciente) y resultados clínicos prometedores, supone una innovación disruptiva en el área clínica de la onco-hematología que viene a posicionarse como una herramienta de gran valor terapéutico en una indicación con escasas posibilidades terapéuticas. La dificultad para realizar comparaciones indirectas con esas alternativas, los restrictivos criterios de inclusión de los ensayos clínicos y su coste económico pueden limitar el acceso al fármaco a determinados pacientes con relativo buen estado funcional. Por tanto, se requieren futuros resultados de eficacia y seguridad a largo plazo para comprender su impacto real y el beneficio de su uso clínico (Fernández-Moriano, 2019b).
—El tratamiento secuencial de la LAL logra en niños unos porcentajes de remisión completa superiores al 90%—
La leucemia mieloide aguda o leucemia aguda mieloide (LMA o LAM) consiste en una proliferación neoplásica de células inmaduras (blastos) de estirpe mieloide, que proceden de un progenitor hematopoyético lesionado con una capacidad de proliferación descontrolada y alterada y que, como consecuencia de su expansión, desplazan la proliferación del resto de células normales de la médula ósea (serie roja, leucocitaria y plaquetaria). La gran diversidad y heterogeneidad de tipos celulares que pueden observarse en la LAM se debe a que la transformación leucémica puede ocurrir en diferentes estadios a lo largo del proceso de diferenciación, lo que condiciona el tipo de células cancerígenas que se podrán encontrar en un determinado paciente. Esto es determinante para su clasificación y pronóstico.
De acuerdo con la última actualización de la clasificación de la OMS en 2016, la LAM y otras neoplasias relacionadas se dividen principalmente en los siguientes grupos:
Globalmente, es el tipo más común de leucemia aguda en adultos (80% de los casos), estimándose una tasa anualizada de incidencia de este grupo de neoplasias en la Unión Europea de alrededor de 5-8 casos por cada 100.000 habitantes al año, y representa un 40% de la mortalidad por leucemias en el mundo occidental, con una tasa anualizada de 4-6 muertes/100.000 habitantes. El registro RETI-SEHOP ha reportado una incidencia global en España en el periodo 2000-2016 de 0,8 casos nuevos por 100.000 niños de 0 a 14 años.
En general, la etiopatogenia de la LAM está en línea con la comentada para la leucemia linfoblástica aguda: se desarrolla como consecuencia de una serie de cambios genéticos en las células precursoras mieloides que regulan la proliferación y la diferenciación, resultando en el acúmulo en la médula ósea de blastos capaces de multiplicarse y proliferar, pero sin la capacidad de diferenciarse a células hematopoyéticas maduras. Entre los posibles factores de riesgo de aparición de LAM en cualquier momento de la vida están: el sexo masculino (proporción 1,3:1 de varones frente a mujeres), el hábito tabáquico (especialmente después de los 60 años), un tratamiento previo con quimioterapia (representa un 10-20% de todos los casos) o radioterapia, haber padecido LAL infantil en el pasado, situaciones de inmunodeficiencia, estar expuesto a la radiactividad o al benceno y tener antecedentes de trastornos hematológicos proliferativos, como el síndrome mielodisplásico; por otro lado, el riesgo de desarrollar LAM es 3 veces mayor entre personas con parentesco en primer grado de pacientes.
De forma similar, la clínica derivada de la LAM se establece de acuerdo a la línea celular que se encuentra disminuida (anemia, neutropenia o trombocitopenia) y/o a la infiltración por células neoplásicas a nivel extramedular (sobre todo, de sistema nervioso central, hígado, bazo, ganglios, piel, pulmones, etc.). El inicio de la enfermedad es relativamente rápido y raramente se superan los 3 meses entre la aparición de los primeros síntomas y el diagnóstico de la LAM. Sin tratamiento precoz, puede avanzar rápidamente y provocar la muerte en un periodo de tiempo corto de semanas o meses.
A la hora de establecer los factores pronósticos, se deben tener en cuenta aquellos que están relacionados con la situación clínica basal del paciente y otros que se relacionan con las características analíticas, citogenéticas y moleculares de la enfermedad. Por tanto, se consideran factores pronósticos desfavorables la edad avanzada, la coexistencia de otras enfermedades, el peor estado clínico del paciente (según escala ECOG, Eastern Cooperative Oncology Group), la hiperleucocitosis, el cariotipo complejo y determinadas alteraciones moleculares. Asimismo, existen clasificaciones pronósticas según las alteraciones citogenéticas/moleculares que estratifican los grupos desde pronóstico favorable (por ejemplo, mutación NPM1) hasta desfavorable (por ejemplo, inversión del cromosoma 3).
El objetivo del tratamiento quimioterápico en pacientes con LAM (Figura 8) es alcanzar la remisión completa y evitar la recidiva. A pesar de los avances en el diagnóstico y la tipificación, el tratamiento de la LAM apenas ha sufrido cambios en las últimas cuatro décadas y, salvo los casos de leucemia promielocítica aguda (un subtipo de LAM que representa una entidad independiente desde el punto de vista terapéutico), comprende, por un lado, la fase orientada a la obtención de la remisión completa (tratamiento de inducción) y, por otro lado, la orientada a evitar la recidiva o tratamiento posremisión (consolidación e intensificación, con o sin trasplante hematopoyético). Por lo general, permite alcanzar tasas de supervivencia a 5 años de casi el 70%: el registro RETI-SEHOP ha estimado un 69% en la cohorte de pacientes de 0-14 años diagnosticados en 2009-2011 en España.
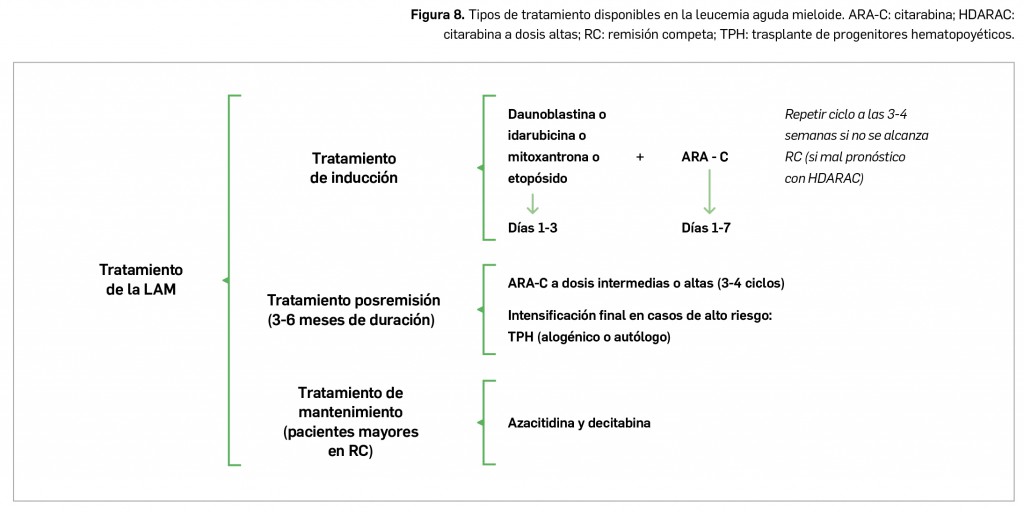
Desde los años 70 del sigo pasado, a fin de reducir la masa leucémica en al menos 3 logaritmos y entrar en “fase invisible”, se ha consolidado como tratamiento estándar citorreductor inicial el clásico esquema 3 × 7, en el que se combinan 3 días de daunorubicina (Daunoblastina®) a 60-90 mg/m2/día con 7 días de arabinósido de citosina –citarabina (Citarabina Accord®, Citarabina Kabi®, Citarabina Pfizer®)– a dosis de 100-200 mg/m2/24 h en infusión continua. Se puede sustituir la daunorubicina por otros inhibidores de la topoisomerasa II como la idarubicina, mitoxantrona y etopósido, con resultados similares. De hecho, la idarubicina o 4’-demetoxidaunorubicina (Zavedos®, Idarubicina Accord® e Idarubicina Sandoz®) a 12 mg/m2/día se ha impuesto como estándar en los últimos años por su mejor penetrabilidad en las células tumorales in vitro, aunque los estudios clínicos comparativos con daunorubicina no han sido del todo concluyentes. Una de las novedades más recientes es que se puede incrementar la dosis de daunorubicina hasta a 90 mg/m2 sin mayor toxicidad, pero con mejor eficacia al compararla con 45 mg/m2, tanto en pacientes mayores como en jóvenes; no está tan clara su superioridad cuando se compara con la dosis de 60 mg/m2.
A la administración de un ciclo de 3 × 7 en inducción le sigue una aplasia de 3-4 semanas de duración, obteniéndose la remisión completa en un 50-60% de los pacientes con un solo ciclo, en cuyo caso la consolidación suele consistir en repetir ese primer ciclo de 3 × 7; no obstante, en la mayoría de los protocolos para LAM de riesgo bajo no se repite la inducción y se procede directamente a intensificación/consolidación con dosis altas de citarabina.
En los casos de persistencia blástica > 10%, particularmente si se confirma fenotipo leucémico por estudios de enfermedad mínima residual, habría 2 opciones. Muchos casos se benefician de administrar inmediatamente un segundo ciclo de inducción 3 × 7 con el que se puede conseguir hasta un 10-20% más de remisión completa. Varios estudios retrospectivos demuestran que la obtención de una buena remisión tras este segundo 3 × 7 no es de especial mal pronóstico. Sin embargo, si tras la quimioterapia de inducción la blastosis medular es > 20%, sobre todo si la LAM mostraba desde el diagnóstico marca-dores de mal pronóstico, en pacientes jóvenes o sin comorbilidad limitante, el segundo ciclo de inducción debe intensificarse utilizando citarabina a dosis altas (HDARAC), combinado o no con un análogo de nucleósidos, con la intención de erradicar la enfermedad residual que persista tras la consolidación.
En conjunto, se considera que la tasa global de remisión completa tras la inducción (con 1 o 2 ciclos) en LAM es del 60-80% y la mortalidad tóxica en inducción del 10-20%. Un 15-20% de pacientes son primariamente resistentes (primary induction failure, PIF) y requieren terapias de inducción de segunda línea con citarabina a dosis altas, o incluso terapias secuenciales con TPH alogénico.
El gran hándicap del abordaje de la LAM es que un 90% de los pacientes en remisión completa recaerá en el plazo de 4-6 meses si el tratamiento no continúa tras la inducción. Cualquier modalidad de terapia posremisión puede prolongar la duración de la remisión, incluyendo la quimioterapia de consolidación o intensificación, mantenimiento o nuevos fármacos. Las mejores tasas de supervivencia libre de enfermedad y una posibilidad de curación del 40-60% en pacientes jóvenes de bajo riesgo se obtienen con una terapia posremisión de 3-6 meses de duración con 3-4 ciclos de máxima intensidad con citarabina a dosis intermedias o altas, seguida de un TPH (alogénico o autólogo) como intensificación final en casos de alto riesgo. La citarabina continúa siendo, pues, la base de la terapia de intensificación en LAM, específicamente en pacientes de riesgo bajo (en menor medida en LAM de cariotipo normal).
Por otra parte, se ha demostrado que la resistencia a la quimioterapia estándar es un factor pronóstico adverso independiente, y también cambia el pronóstico inicial. Por tan-to, hoy en día es imprescindible practicar estudios genéticos amplios y un seguimiento seriado de la enfermedad mínima residual tras las consolidaciones, ya que muchos pacientes de buen pronóstico, pero con leucemia residual tras inducción y consolidación, se beneficiarían de un TPH alogénico antes de perder la primera remisión completa.
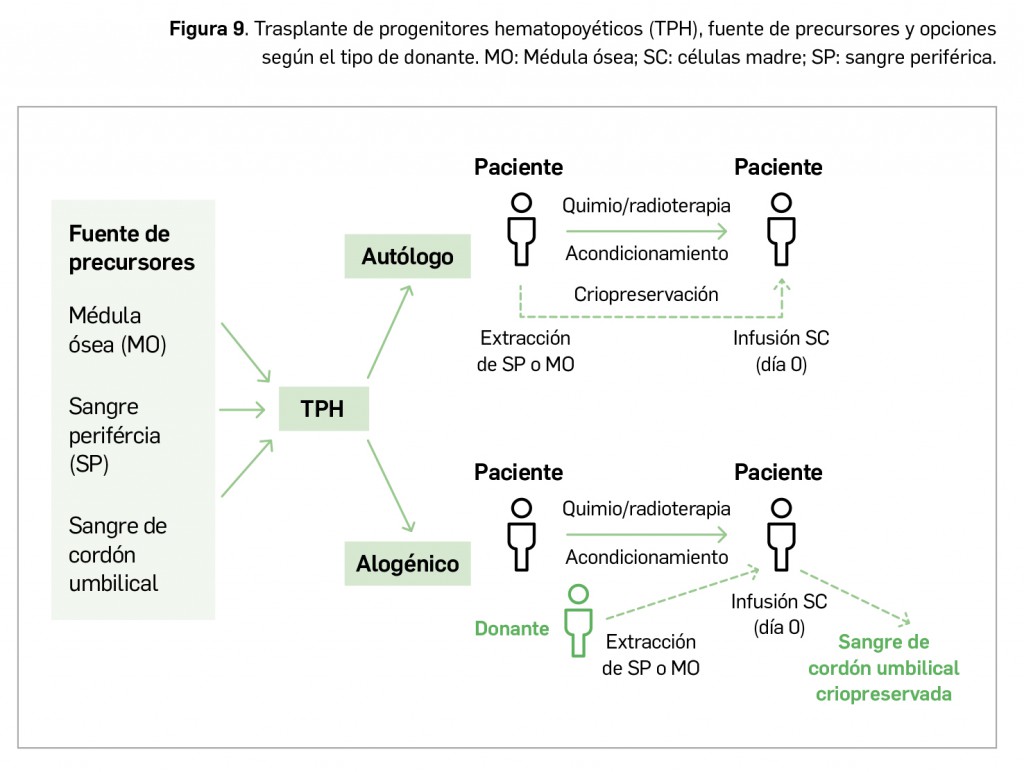
A grandes rasgos, el TPH (Figura 9) se considera una forma de intensificación en primera remisión completa para lograr la curación en LAM, y aún hoy en día la LAM de alto riesgo es la indicación más frecuente de trasplante alogénico en el mundo. Según los donantes, el TPH puede ser autólogo, alogénico familiar de hermano HLA idéntico o haploidéntico (TPH-Haplo) o no familiar de donante no emparentado voluntario (TPH-DNE). La fuente puede ser la médula ósea o los precursores movilizados a sangre periférica en adultos, o el trasplante de cordón umbilical alogénico (TPH-SCU). La intensidad del TPH se define según el acondicionamiento sea mieloablativo o de intensidad variablemente reducida (Tabla 3).
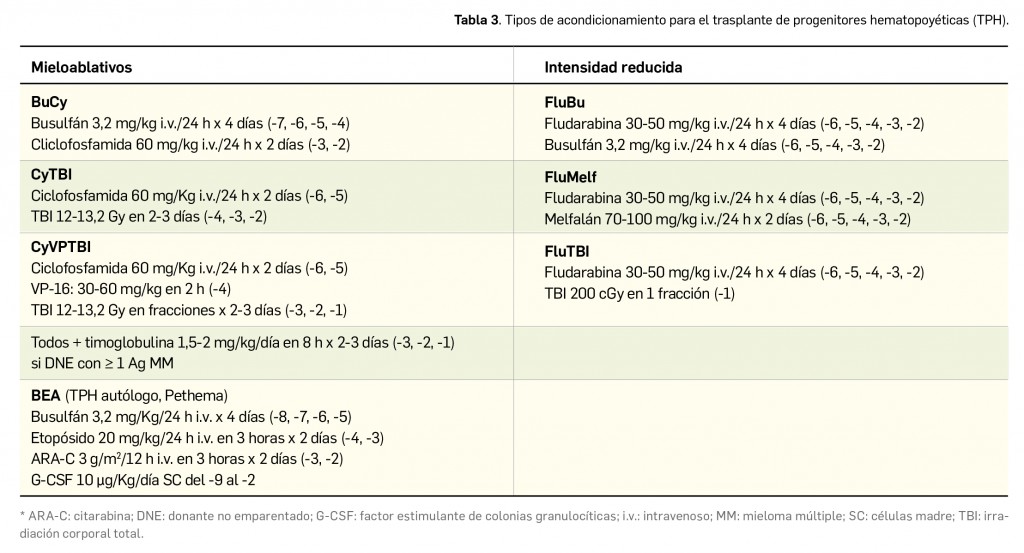
El trasplante autólogo carece de efectos aloinmunes y, por tanto, toda su eficacia se basa en la potencia antileucémica del acondicionamiento. Por su parte, el alogénico cuenta además con el efecto injerto contra leucemia ejercido por las células efectoras inmunes del donante. El trasplante puede efectuarse en distintos momentos de la terapia de la LAM, a pacientes con un amplio espectro de edades y situación basal. La suma de las múltiples variables del procedimiento, de la leucemia y su respuesta a la terapia, y de las circunstancias del propio paciente genera un complejo sistema de interacciones que hay que valorar cuidadosamente para elegir lo más adecuado para cada caso, complicando enormemente las decisiones terapéuticas.
Excepto en la leucemia promielocítica aguda, el mantenimiento con quimioterapia no ha demostrado utilidad en LAM cuando se compara con la terapia habitual de inducción y consolidación referida previamente en pacientes jóvenes. En cambio, es bastante probable que funcionen las terapias con hipometilantes lentos, como azacitidina (Vidaza® y EFG) y decitabina (Dacogen®), en adultos mayores en remisión completa con enfermedad controlada, pero no candidatos a continuar con la quimioterapia.
Con los esquemas de tratamiento actualmente disponibles, la LAM en pacientes no pediátricos tiene una supervivencia al año de solo el 50% y a los 5 años del 19-28%, habiéndose alcanzado un tope de curación inferior al 50% para los adultos jóvenes y menor del 10% para los pacientes mayores de 60 años no aptos para recibir tratamientos intensivos. Por ello, y especialmente para los casos primarios, sigue existiendo una clara necesidad de nuevos tratamientos con terapias dirigidas frente a nuevas dianas moleculares que mejoren los resultados ofrecidos por la quimioterapia (Alegre et al., 2017).
A este respecto, recientemente ha habido dos interesantes innovaciones terapéuticas. La primera de ellas, la midostaurina (Rydapt®), para pacientes con mutaciones en el gen FLT37 (13q12), una de las más relevantes identificadas en la LAM con cariotipo normal y que está presente en aproximadamente un 30% de los casos de nuevo diagnóstico en adultos, asociada con un peor pronóstico: tasas de recaída de hasta el 85% a los 5 años del diagnóstico y supervivencias más reducidas (15% a 5 años). Aproximadamente tres cuartas partes de los pacientes con mutaciones en el gen FLT3 tienen una mutación tipo duplicación interna en tándem de aminoácidos dentro del dominio yuxtamembrana del receptor (FLT3-ITD o internal tandem duplication mutation), las cuales se identifican en el 20-30% de los pacientes adultos con LAM y se asocian con un peor pronóstico. En algo menos del 10% de los pacientes se identifica el subtipo FLT3-TKD (Tyrosine Kinase Domain), asociado a mutaciones puntuales en el dominio de la segunda tirosina cinasa y cuyo pronóstico es controvertido pero más favorable (Fathi et al., 2017).
La midostaurina es un nuevo fármaco inhibidor de múltiples tirosina cinasas que aporta cierta innovación a nivel mecanístico ya que, a pesar de ser un inhibidor no-selectivo de estas enzimas, como otros muchos fármacos disponibles en España, incorpora una mayor afinidad sobre los receptores FLT3 y KIT, ausentes del perfil farmacodinámico de otros fármacos, que resulta determinante en sus efectos biológicos. Al unirse a los dominios catalíticos de dichos receptores, inhibe la transducción de señales mediada por ellas: detiene el ciclo celular e induce la apoptosis de células leucémicas. El medicamento, designado como huérfano, ha sido oficialmente autorizado para el tratamiento por vía oral de pacientes adultos con LAM de nuevo diagnóstico con mutación FLT3, en combinación con quimioterapia de inducción (daunorubicina y citarabina) y de consolidación (altas dosis de citarabina) seguido de tratamiento de mantenimiento en monoterapia en pacientes que han alcanzado respuesta completa. Adicionalmente, se ha aprobado para el tratamiento en monoterapia de pacientes adultos con mastocitosis sistémica agresiva, mastocitosis sistémica con neoplasia hematológica asociada o leucemia mastocítica.
Los datos de seguridad y eficacia clínica en LAM proceden de un amplio ensayo de fase 3 (N= 717) en pacientes adultos con mutación FLT3, en que midostaurina (asociado a un régimen de quimioterapia estándar) redujo el riesgo de muerte en un 23% frente al brazo de placebo, prolongando significativamente la mediana de supervivencia global hasta 74,7 meses (vs. 25,6 meses con placebo) e incrementando la supervivencia a largo plazo (5 años) en un 7-8% (51% vs. 44% con placebo). El beneficio clínico –que se considera relevante por el mal pronóstico de estos pacientes y las escasas opciones terapéuticas– se mantenía independientemente de que recibieran TPH, y se confirma por los resultados de variables secundarias como la supervivencia libre de eventos (8,2 vs. 3,0 meses) y la tasa de remisión completa (59% vs. 54%).
El perfil de seguridad de midostaurina en periodos de exposición cercanos a 1 año parece, en líneas generales, aceptable (tasa de interrupción cercana al 3%) y manejable; aunque muchos pacientes experimentan eventos adversos graves (incidencia similar a placebo), la mayoría de ellos se atribuyen a la quimioterapia concomitante. Las reacciones adversas más comunes con el tratamiento con midostaurina derivan de la toxicidad gastrointestinal y hematológica, manifestada como náuseas y vómitos, y citopenias. Por su gravedad (grado ≥ 3) destacan la neutropenia febril (83%), linfopenia (20%), infección relacionada con el dispositivo (16%), dermatitis exfoliativa (14%, ligeramente superior a placebo), hiperglucemia (7%) y náuseas (6%). La evaluación de las dos fases de tratamiento por separado sugirió que la incidencia global de eventos adversos durante el mantenimiento fue menor que durante la fase de inducción/consolidación, en la que se reportaron la mayoría de las alteraciones hematológicas graves (Fernández-Moriano, 2019c).
Así pues, si bien no representa una cura y se requieren futuros estudios para esclarecer algunas incertidumbres sobre su eficacia y seguridad (como el beneficio-riesgo en pacientes de > 60 años), midostaurina se posiciona como alternativa útil en primera línea, y quizá de primera elección para formar parte del tratamiento estándar de pacientes adultos con LAM con mutación FLT3.
El otro nuevo fármaco incorporado en 2019 a la terapéutica de la LAM es gemtuzumab ozogamicina (Mylotarg®). Con un fundamento similar al ya comentado inotuzumab ozogamicina, se trata de un conjugado compuesto por el anticuerpo monoclonal humanizado anti-CD33, gemtuzumab, enlazado covalentemente con la molécula pequeña N-acetil-γ-calicheamicina, un producto natural semisintético citotóxico. Así, se dirige específicamente contra las células que expresan CD33 en su superficie, como los linfoblastos leucémicos mieloides, y tras su internalización, libera el derivado de calicheamicina, cuya activación induce roturas del ADN bicatenario de la célula cancerígena, la interrupción del ciclo y la muerte celular por apoptosis. El medicamento, designado como huérfano, ha sido oficialmente autorizado para el tratamiento combinado con daunorubicina y citarabina en el tratamiento de pacientes a partir de los 15 años de edad con leucemia mieloide aguda CD-33+ de novo no tratada previamente, excepto la leucemia promielocítica aguda.
La eficacia del fármaco en la indicación y pauta autorizadas ha sido adecuadamente contrastada en un amplio ensayo pivotal de fase 3 (N= 271), multicéntrico, aleatorizado, controlado y abierto. La adición de dosis fraccionadas –por vía intravenosa– de gemtuzumab ozogamicina a la quimioterapia estándar (esquema 3+7 de daunorubicina más citarabina) demostró un aumento estadísticamente significativo de 7,8 meses en la mediana de supervivencia libre de eventos (SLE) respecto al tratamiento con la quimioterapia exclusivamente (17,3 meses vs. 9,5 meses; HR= 0,56; p= 0,0002), con una reducción del 44% del riesgo de eventos en pacientes naïve de 50-70 años. La supervivencia libre de recaídas aumentó en casi 17 meses (28,0 vs. 11,4 meses; HR= 0,53; p= 0,0006), lo cual corrobora la prolongación de la remisión con la combinación. No obstante, el número de pacientes que alcanza la remisión no varía (no hubo cambios significativos en la tasa de respuesta objetiva) y los resultados de supervivencia global, aunque con tendencia favorable para la combinación, no son concluyentes (mediana de 21,8 vs. 27,5 meses; HR= 0,81; p= 0,1646).
Adicionalmente, otro estudio aleatorizado y controlado (AAML0531; Cooper et al., 2012) investigó el efecto de gemtuzumab ozogamicina en población pediátrica con LAM. Incluyó a un total de 1.022 niños y adultos jóvenes recién diagnosticados (>94% menores de 18 años; mediana de edad 9,7 años), de los cuales 350 pacientes recibieron el fármaco en combinación con la quimioterapia de inducción y consolidación. Aunque la tasa de remisión no se veía mejorada –pero se mantenía en niveles altos de remisión completa (de hasta el 87%)–, el riesgo de recaídas se vio significativamente reducido con la adición del fármaco, con mejoría de la estimación de SLE a los 3 años hasta el 53,1% (vs. 46,9% para la quimioterapia sola; HR= 0,83; IC95% 0,70-0,99; p= 0,04); la tendencia era también hacia una mayor SG a los 3 años (69,4% vs. 65,4%), aunque los resultados no alcanzaban significación estadística (HR= 0,91; IC95% 0,74-1,13; p= 0,39).
Por otra parte, el perfil toxicológico de gemtuzumab ozogamicina parece aceptable, esperable en base al mecanismo de acción del fármaco y clínicamente manejable, si bien la asociación del fármaco a la quimioterapia aumenta la incidencia de eventos adversos graves relacionados con el tratamiento (87,0% vs. 75,9% en el brazo control) y de interrupción del tratamiento por los mismos (29,0% vs. 2,9%). Las infecciones y las hemorragias graves destacan como las reacciones adversas más frecuentes, siendo los trastornos hepatobiliares venooclusivos y la trombocitopenia motivos importantes de abandono del tratamiento. La toxicidad del fármaco se reveló mayor en los pacientes pediátricos, con una mortalidad posremisión incrementada que se atribuyó a la neutropenia prolongada.
Así pues, el positivo balance beneficio-riesgo de gemtuzumab ozogamicina sugiere que el fármaco se va a sumar al régimen estándar de tratamiento (daunorubicina con citarabina) para constituir un nuevo “standard of care” en primera línea de los casos de LAM (no promielocítica), independientemente del estado mutacional del gen FLT3. Asumiendo que tampoco representa un tratamiento curativo, el beneficio que aporta este fármaco frente a la quimioterapia estándar, aunque moderado, es clínicamente relevante si se considera el alto riesgo de recaídas en la LAM y el pobre pronóstico de supervivencia y la ausencia de alternativas terapéuticas eficaces en casos de enfermedad recidivante. Los pacientes con un riesgo citogenético favorable o intermedio –suponen el 70-80% de los pacientes de nuevo diagnóstico– podrán beneficiarse en mayor medida de la prolongación de la remisión. El fármaco abrió una nueva vía en cuanto a mecanismo de acción en el tratamiento de la enfermedad (Fernández-Moriano, 2019d).
Los linfomas de Hodgkin (LH) son tumores poco frecuentes que consisten en una proliferación, localizada o diseminada, de células tumorales que se originan en el sistema linforreticular y que afecta principalmente los ganglios linfáticos y la médula ósea. El LH es, en realidad, un grupo heterogéneo de neoplasias linfoides malignas de origen mayoritario en linfocitos B del centro germinal, que se caracteriza histológicamente por la presencia de células de Hodgkin y de Reed-Sternberg (HRS). No obstante, en el ganglio linfático de estos pacientes las células HRS representan solo una pequeña proporción (5%) de las células que ocupan el ganglio; el resto del ganglio está ocupado por un infiltrado inflamatorio formado básicamente por linfocitos T y otras células como eosinófilos, macrófagos, etc. Teóricamente, todas aquellas zonas del organismo que contienen tejido linfoide pueden verse afectadas (hígado, bazo, médula ósea, amígdalas, etc.), aunque esta afectación extraganglionar es poco común, al menos en el momento del diagnóstico de la enfermedad.
El linfoma se forma a partir de un linfocito que se encuentra en proceso de maduración/activación en el ganglio linfático. En condiciones normales, si dicho linfocito sufre una mutación en los genes que regulan su capacidad para producir anticuerpos, transformándose en una célula incapaz de llevar a cabo su función fisiológica, experimentará un proceso de apoptosis. Sin embargo, algunas mutaciones determinan la aparición de resistencia frente a los mecanismos apoptóticos naturales encargados de eliminar las células que acumulan múltiples deficiencias, mientras otras mutaciones permiten escapar al control de las células encargadas de la vigilancia inmunológica y proliferar de forma incontrolada. Por lo general, el LH comienza a afectar a los ganglios linfáticos del cuello o del mediastino (entre los pulmones y la zona posterior del esternón). También pueden aparecer en los grupos de ganglios linfáticos que están en las axilas, en las ingles, en el abdomen o en la pelvis. En el caso de que se produzca la diseminación de los linfocitos mutados, lo más frecuente es que lo hagan al bazo o al hígado, aunque también pueden diseminarse –con mucha menor frecuencia– al pulmón, hueso y médula ósea.
El linfoma de Hodgkin se presenta bajo dos formas clínicas principales:
El LH tiene una incidencia global de 4-5 casos/100.000 habitantes/año, representando el 20-25% de todos los linfomas. Según el Instituto Nacional del Cáncer (NIH) estadounidense, en 2018 los linfomas de Hodgkin constituían el 0,5% de todos los nuevos casos y un 0,2% de los fallecimientos por cáncer en EE.UU., con tasas de supervivencia a 5 años del 87%. La mayoría de los pacientes se diagnostican entre los 15 y los 30 años de edad, con otro pico de incidencia en pacientes mayores de 55 años. El LH es algo más frecuente en varones, si bien el LHc de subtipo esclerosis nodular y los casos que debutan en el primer pico de edad predominan en mujeres.
No parece existir una clara asociación de factores familiares y socioeconómicos con la aparición de la enfermedad, aunque en el caso de los adolescentes en países occidentales, la enfermedad afecta más frecuentemente a individuos que pertenecen a familias de mayor nivel socioeconómico (Sureda, 2020). En niños es una enfermedad poco frecuente, que representa aproximadamente el 5% de los cánceres infantiles. El registro RETI-SEHOP reporta una incidencia en España de 0,74 casos nuevos por 100.000 en niños de 0 a 14 años en el periodo 2000-2016.
En líneas generales, la etiopatogenia del LH es desconocida; pero parece que están implicados factores tanto inmunológicos como genéticos y ambientales. Ciertas peculiaridades epidemiológicas sugieren la posibilidad de que exista algún microorganismo implicado en la etiopatogenia de la enfermedad. Uno de los más frecuentemente asociados es el virus de Epstein-Barr (VEB)8, dado que presenta una tendencia natural a infectar a los linfocitos B. A este respecto, haber padecido una mononucleosis infecciosa triplica la probabilidad de padecer un LH, e incluso se ha detectado material genético del VEB en el interior de los linfocitos B malignos del LH en aproximadamente la mitad de los casos (hasta en el 75% del subtipo celularidad mixta); no obstante, esta observación es más común en los países en vías de desarrollo (> 90% de los casos) que en los países desarrollados (40%). Según los datos actualmente disponibles, el material genético aportado por el VEB podría colaborar en los mecanismos por los que la célula maligna elude los mecanismos fisiológicos apoptóticos antes de desarrollar el linfoma.
Asimismo, hay datos epidemiológicos que apuntan a la posibilidad de una cierta predisposición genética para desarrollar un LH. De hecho, los familiares en primer grado de los pacientes presentan un riesgo hasta 5 veces mayor de sufrir el linfoma y los gemelos monocigóticos de pacientes con LH tienen una probabilidad casi 100 veces mayor de padecer la enfermedad en relación a los gemelos dicigóticos. En cualquier caso, la probabilidad de una agregación familiar hereditaria (LH familiar) es inferior al 5% de todos los casos.
Desde el punto de vista clínico, hay que subrayar que en el 60-70% de los casos el paciente se encuentra asintomático en el momento del diagnóstico, y consulta por crecimiento ganglionar o por tos persistente seca que conduce al hallazgo de una masa mediastínica en una radiografía de tórax. Según se ha indicado, los ganglios tumorales inflamados se suelen localizar preferentemente en la regiones cervical y supraclavicular (60-80% de casos), sobre todo izquierdas, confundiéndose en muchas ocasiones con ganglios inflamados secundarios a infecciones bucales, dentarias o de oídos; otras veces, estos ganglios aumentados de tamaño se localizan en región axilar (10-20%) o inguinal (6-12%), siendo también frecuente la afectación mediastínica en el subtipo de esclerosis nodular. Las adenopatías son de consistencia elástica y generalmente indoloras, aunque en el 10% de los casos –principalmente en pacientes jóvenes– la ingesta de alcohol induce dolor intenso en las adenopatías afectas (signo de Oster). Adicionalmente, en aproximadamente 1 de cada 3 casos, se detecta esplenomegalia al diagnóstico, pero el éste debe de confirmarse mediante la biopsia de un ganglio linfático sospechoso.
Otras manifestaciones habituales del LH suelen ser:
El sistema de clasificación del LH de Ann Arbor establece diferentes estadios dependiendo del grado de afectación:
I: afectación de una sola región de ganglios linfáticos o de un solo órgano o sitio extralinfático.
II: afectación de 2 o más regiones de ganglios linfáticos en el mismo lado del diafragma, o bien afectación localizada de un solo órgano o sitio asociado extralinfático y sus ganglios linfáticos regionales.
III: afectación de ganglios linfáticos en ambos lados del diafragma que también puede ir acompañada de la afectación localizada de un órgano o sitio extralinfático, de la complicación del bazo, o de ambos.
IV: afectación diseminada de uno o más sitios extralinfáticos con asociación de ganglio linfático o sin ella, o con afectación aislada de un órgano extralinfático y complicación ganglionar distante (no regional).
El índice pronóstico internacional (IPS, international prognostic score) utiliza un conjunto de 7 factores de riesgo en el momento del diagnóstico: sexo varón, > 45 años de edad, enfermedad en estadio IV, albúmina sérica < 4 g/dl, hemoglobina < 10,5 g/l, recuento de leucocitos > 15.000/mm3) y recuento de linfocitos < 600/mm3 o < 8% del recuento total de leucocitos. Cada factor puntúa 1 y se considera que un IPS > 4 es desfavorable.
Como en algunos otros tumores hematológicos, el objetivo del tratamiento del linfoma de Hodgkin es conseguir la mayor curación de pacientes con la menor toxicidad aguda y a largo plazo. Para ello, es fundamental adecuar el tratamiento al paciente y a las características del LH (estadio, factores pronósticos y respuesta al tratamiento). A grandes rasgos, el tratamiento del LH está basado en la utilización de poliquimioterapia en ocasiones asociada a radioterapia sobre campo afecto, quimioterapia a altas dosis con TPH autólogo y, más recientemente, se han ido incorporando nuevos fármacos biotecnológicos que se agrupan dentro de la inmunoquimioterapia.
Si no existen factores adversos y se administra el tratamiento adecuado, el LH se considera actualmente una enfermedad curable en al menos el 75-80% de los pacientes (90% para los primeros estadios I/II y del 70% para los estadios avanzados III/IV en el momento del diagnóstico). En población pediátrica, se ha estimado una supervivencia a 5 años del 97% en niños de 0 a 14 años diagnosticados en la cohorte de 2009-2011.
No obstante, a pesar del buen pronóstico generalizado, los enfermos que no consiguen una remisión completa tras el tratamiento inicial (5-15%) o los pacientes con recidivas tardías posteriores a una remisión completa (30%) tienen un pronóstico mucho más desfavorable. Ambos tipos de pacientes serán tratados con esquemas de quimioterapia de segunda línea, de carácter más intensivo que los de primera y, sobre todo, con algunos fármacos diferentes a los empleados en primera línea. Si el paciente consigue una respuesta inicial, está indicado consolidarla con quimioterapia a dosis altas y trasplante autólogo de progenitores hematopoyéticos (éste no se utiliza como el primer tratamiento para el LH, pero puede recomendarse en pacientes con formas refractarias/recurrentes). Los pacientes que recaen después del TPH autólogo tienen peor pronóstico.
Una excepción la constituye la forma de LH de predominio linfocítico nodular, para la que se dispone de datos contrastados que indican que algunos pacientes mejoran con solo la escisión quirúrgica de la masa tumoral, cuando ello es posible. Además, cuando recaen, las recaídas no suelen ser tan agresivas y las tasas de supervivencia son un poco mayores que las de la forma clásica de LH.
Este tratamiento fue el primero que consiguió curar a algunos pacientes con estadios iniciales, pero su uso ha ido decayendo a favor de la quimioterapia. De hecho, actualmente la radioterapia se utiliza casi siempre combinada con quimioterapia, empleando la radioterapia de campo afectado, mediante la que se irradian exclusivamente las áreas de los ganglios linfáticos afectados. La modalidad de tratamiento combinado de radioterapia y quimioterapia sistémica es el tratamiento estándar para los pacientes con LH en estadio inicial, pues es altamente eficaz y conlleva un muy buen pronóstico, aunque es necesario un seguimiento prolongado con exámenes físicos, test sanguíneos y pruebas de imagen para detectar posibles recaídas y los efectos secundarios a largo plazo de los tratamientos citotóxicos utilizados.
Los efectos secundarios inmediatos producidos por la radioterapia dependen del área del cuerpo que se está tratando, y pueden incluir: astenia, reacciones leves en la piel, malestar estomacal y deposiciones líquidas; los pacientes que recibieron radioterapia en el cuello quizá tengan dolor en la boca y/o la garganta. Si bien el riesgo de efectos secundarios a largo plazo ha disminuido con las mejoras en el tratamiento, la radioterapia aún puede causar ciertos problemas de toxicidad, que incluyen cánceres secundarios, infertilidad, inmunidad reducida, problemas de tiroides, enfermedad cardiaca y accidentes cerebrales vasculares.
Es precisamente el riesgo de efectos adversos a largo plazo, como el desarrollo de neoplasias malignas secundarias, el motivo por el cual se ha cuestionado el beneficio de la radioterapia, y algunos grupos de estudio apoyan la quimioterapia sola para esta indicación. Sin embargo, los resultados de un meta-análisis reciente (Blank et al., 2017), que ha comparado la combinación de quimioterapia sola y quimioterapia más radioterapia, con el mismo número de ciclos de quimioterapia en ambos brazos, no parece apoyar la preferencia de la quimioterapia sola sobre la combinación con radioterapia. De hecho, la adición de radioterapia a la quimioterapia mejoró la supervivencia global en comparación con la quimioterapia sola (cocientes de riesgo instantáneo o hazard ratio, HRi= 0,31; IC95% 0,19-0,52; p< 0,00001); asimismo, la administración de quimioterapia y radioterapia mejoró la supervivencia libre de progresión (HRi= 0,42; IC95% 0,25-0,72; p= 0,001). En relación a la mortalidad relacionada con infecciones sobrevenidas (HR= 0,33; IC95% 0,01-8,06; p= 0,5), a un segundo cáncer (HR= 0,53; IC95% 0,07-4,29; p= 0,55) y a enfermedades cardiacas (HR= 2,94; IC95% 0,31-27,55; p= 0,35), no hay evidencia de una diferencia estadísticamente significativa entre la administración de quimioterapia sola y quimioterapia más radioterapia. Para la tasa de respuesta completa, tampoco se encontraron diferencias significativas entre los grupos de tratamiento (HR= 1,08; IC95% 0,93-1,25; p= 0,33).
Los regímenes de quimioterapia, asociados o no a radioterapia, implican la administración de diferentes fármacos antineoplásicos cada 2 a 3 semanas, generalmente de forma escalonada. En caso de alcanzar una respuesta completa o parcial, el paciente podrá ser sometido a quimioterapia a altas dosis, seguida de un TPH autólogo con células madre de médula ósea que se extraen cuando el paciente está en remisión de la enfermedad después del tratamiento previo; de lo contrario, deberá recibir algún otro esquema. Si tras el TPH autólogo recae, se deberá considerar la opción de un TPH alogénico con células de un donante compatible (generalmente, un familiar próximo), a fin de que el sistema inmunitario del donante destruya las células cancerosas del paciente.
En primera línea, las combinaciones más ampliamente utilizadas son: ABVD –doxorubicina, bleomicina, vinblastina y dacarbazina–, que generalmente se administra cada 2 semanas durante 2 a 8 meses, y BEACOPP –bleomicina, etopósido, doxorubicina, ciclofosfamida, vincristina, procarbazina y prednisona–. Un meta-análisis (Skoetz et al., 2017) con datos de 4 ensayos clínicos que incluyeron a más de 2.800 pacientes ha concluido que el régimen de BEACOPP escalonado aumenta la supervivencia global (HR= 0,74; IC95% 0,57 a 0,97) en relación al de ABVD; en concreto, se observó una reducción del 25% en el número de pacientes que morirán después de 5 años en el brazo de BEACOPP escalonado en comparación con el de ABVD. Esta ventaja de supervivencia también se refleja en una mayor supervivencia libre de progresión con BEACOPP escalonado (cociente de riesgos instantáneos, HRi= 0,54; IC95% 0,45-0,64), lo cual significa que después de 5 años el número de pacientes que experimentarán un progreso de la enfermedad, recidiva o la muerte en el brazo de BEACOPP escalonado será un 42% inferior en comparación con el de ABVD.
Otros regímenes quimioterápicos empleados en primera línea, aunque con menor frecuencia, son:
En segunda línea, muchos de estos tratamientos se brindan en preparación para un trasplante autólogo, pero también para controlar la enfermedad y sus síntomas. Los esquemas quimioterápicos de rescate más utilizados en caso de recaída, por lo general durante 2 semanas consecutivas seguidas de una semana libre, o cada 2 semanas, son:
En general, los efectos secundarios de la quimioterapia convencional dependen del paciente y de la dosis utilizada, pero pueden incluir: riesgo de infección, náuseas y vómitos, neuropatía periférica, caída del cabello, pérdida del apetito y estreñimiento. En los pacientes tratados con protocolos de quimioterapia intensificados, se observa, además, a cambio de una mayor eficacia, un aumento en el riesgo de leucemia mieloide aguda y de síndrome mielodisplásico secundarios, lo que hace necesario individualizar los tratamientos.
Una opción un tanto especial en segunda línea, aunque solo para pacientes adultos, la constituye el brentuximab vedotina (Adcetris®, comercializado en 2014): un conjugado que consta de un anticuerpo anti-CD30 (brentuximab) capaz de dirigir el agente quimioterápico (vedotina) a las células que expresan en su superficie la proteína CD309. El anticuerpo se une selectivamente a la parte extracelular de CD30 presente en la superficie de la membrana, formando un complejo que es internalizado mediante endocitosis. Una vez en el interior celular, el fármaco accede al interior de los lisosomas donde sufre un proceso de escisión proteolítica liberando la vedotina (monometilauristatina E), un potente agente citotóxico que actúa interfiriendo con la polimerización de la tubulina e impide la formación del huso mitótico durante la división celular, bloqueando el ciclo celular en fase G2/M y provocan la activación de los mecanismos de apoptosis (muerte celular programada) y, en definitiva, la muerte celular (AEMPS, 2020).
El medicamento ha sido autorizado para el tratamiento de pacientes adultos con LH CD30+ en recaída o refractario después de TPH autólogo (o si tras éste tienen alto riesgo de recaída/progresión) o tras ≥ 2 tratamientos previos cuando el TPH autólogo o la poliquimioterapia no es una opción terapéutica. Generalmente, se administra cada 3 semanas durante un máximo de 16 ciclos, aunque a veces se administra cada 4 semanas. Se está evaluando como un complemento o reemplazo de la quimioterapia antes del trasplante para los pacientes con LH recurrente.
Aunque aún no se ha establecido su seguridad y eficacia en población pediátrica, en el último lustro se han incorporado dos opciones en este campo para el tratamiento del LH, como son nivolumab (Opdivo®) y pembrolizumab (Keytruda®), que inauguraron una vía terapéutica de gran relevancia en oncología. Además de para otras indicaciones, están autorizado en monoterapia para el tratamiento de pacientes adultos con LH clásico en recaída o refractario después de un TPH autólogo y de tratamiento con brentuximab vedotina, o no candidatos a éstos. Ambos son anticuerpos monoclonales enteramente humanos de la clase IgG4 que se unen selectivamente y con gran afinidad a la proteína de muerte programada PD-1 (programmed death 1; CD279), un receptor de superficie celular que desempeña un papel importante en la regulación negativa del sistema inmunitario y la promoción de la auto-tolerancia mediante la supresión de la actividad inflamatoria de las células T.
De hecho, PD-1 constituye un punto de control que protege contra la autoinmunidad a través de un mecanismo dual de promoción de la apoptosis en linfocitos T específicos de antígeno en los ganglios linfáticos y, simultáneamente, de reducción de la apoptosis en linfocitos T reguladores (de carácter inmunosupresor, por reducir la inducción y proliferación de los linfocitos T efectores). Sobre la PD-1 actúan sus ligandos naturales PD-L1 y PD-L2, que son expresados en diversos tipos de células tumorales, así como en las células presentadoras de antígenos (CPA), linfocitos B y linfocitos T activados; su unión al receptor provoca la inhibición de la proliferación y de la secreción de citocinas de los linfocitos T efectores. Es preciso tener en cuenta que el balance de la inmunidad mediada por las células T es determinante en el control de enfermedades infecciosas y del cáncer, así como en el desarrollo de tolerancia inmunológica a los antígenos propios (a través de las CPAs). La inducción y el mantenimiento de la tolerancia mediada por células T a través de la vía PD-1 limita la respuesta de subpoblaciones de células T efectoras para evitar que se produzca un daño tisular como resultado de una excesiva actividad inmunitaria. En definitiva, la función fisiológica de la vía PD-1 consiste en modular la actividad de las células T para asegurar que el sistema inmunitario no permanezca siempre «encendido» (en estado de hiperactivación) después de cualquier infección viral o bacteriana.
Conviene subrayar que las alteraciones en la interacción de PD-1 y sus ligandos PD-L1 y PD-L2 son utilizadas como un mecanismo de escape inmunológico por parte de las células tumorales, facilitando la proliferación y la distribución distal de éstas. De forma similar, ciertos microorganismos pueden alterar esta vía dando como resultado la persistencia de las infecciones crónicas. En base a lo anterior, la inhibición del receptor PD-1 potencia la respuesta antitumoral “natural” de los linfocitos T, incluyendo la producción de citocinas por parte de éstos. Esta estrategia se ha revelado eficaz frente a diversos tipos de cáncer.
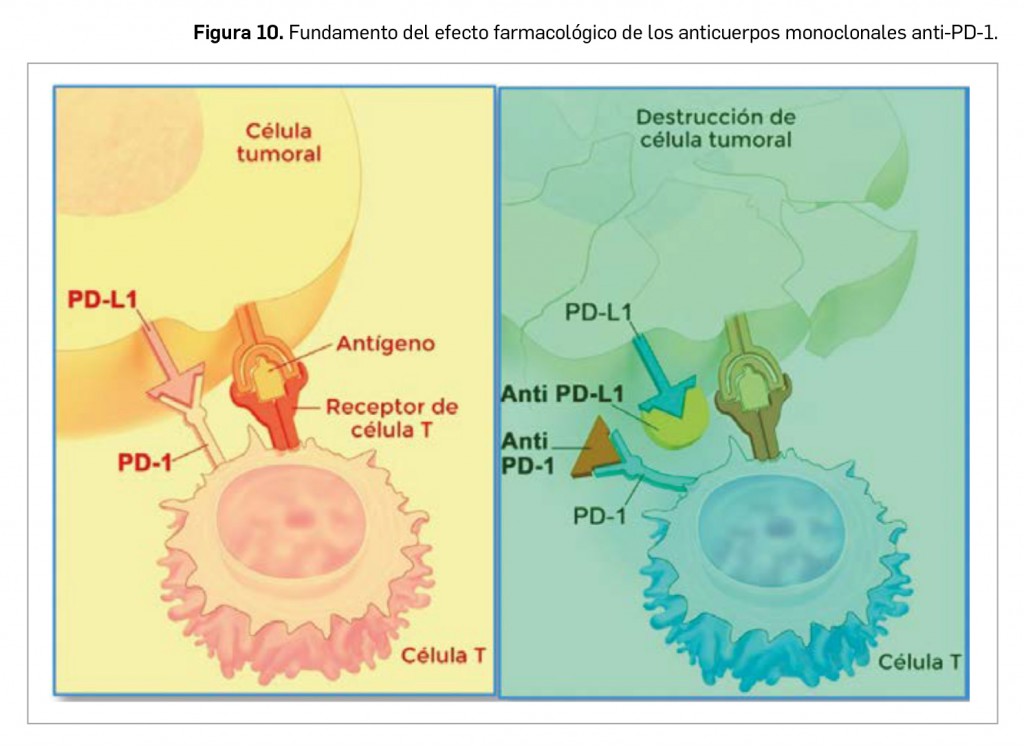
Las redes inmunosupresoras en los linfomas van desde cambios en la composición del microambiente celular hasta distintas vías de señalización, como PD-1/PD-L1. Si bien el ejemplo prototípico de un linfoma que manipula y silencia el sistema inmunitario es el LH, también otros linfomas, como el de células B mediastínico primario y algunas neoplasias malignas del virus de Epstein-Barr (VEB), utilizan estrategias de supervivencia análogas, mientras que el linfoma difuso de células B grandes, linfoma folicular o el linfoma T angioinmunoblástico ejercen estrategias de escape inmunológico adicionales (Menter et al., 2018).
El LH clásico se caracteriza por alteraciones genéticas casi universales en 9p24.1, que dan como resultado la expresión constitutiva de los ligandos de PD-1. Esto justifica la gran sensibilidad de los LH al bloqueo PD-1, con tasas de respuesta del 70% en la enfermedad recurrente/refractaria. En la actualidad hay numerosos ensayos clínicos en curso que están probando el uso de inhibidores de la PD-1 en etapas más tempranas del tratamiento y en combinación con diversas terapias. Por su parte, los linfomas no-Hodgkin (LNH) no muestran alteraciones de 9p24.1 y, por tanto, no comparten la vulnerabilidad de los LH al bloqueo de PD-1; sin embargo, algunos subtipos de LNH se salen de la norma y muestran características genéticas o inmunológicas que pueden predecir la sensibilidad al bloqueo de esta vía, como el linfoma primario de células B mediastínicas, el linfoma primario del sistema nervioso central y el linfoma testicular primario, que sí albergan alteraciones frecuentes en 9p24.1, o los linfomas por infección por el VEB, que aumenta la expresión de PD-L1 (Menter et al., 2018).
Conviene recordar que, como se ha observado con otras opciones de inmunoterapia, los efectos antitumorales de nivolumab y pembrolizumab podrían manifestarse de forma retardada (en torno a 2-3 meses) en relación a la quimioterapia estándar, cuyo efecto citotóxico es prácticamente inmediato. Este retardo en la respuesta debe ser considerado antes de iniciar el tratamiento en pacientes cuya enfermedad neoplásica progresa rápidamente o en los que la expectativa de vida sin tratamiento no sea superior a 3 meses.
Por otra parte, el uso inicial de anticuerpos anti-CD20, como rituximab o ofatumumab, en linfoma de Hodgkin todavía es controvertido, ya que algunos indicios apuntan a que podrían desencadenar una enfermedad más agresiva; sin embargo, los pacientes en recaída tras la quimioterapia ABVD pueden alcanzar remisiones hasta del 80% al ser tratados con regímenes combinados con anti-CD20 (Cuéllar, 2018).
Los linfomas no-Hodgkin (LNH) son un grupo heterogéneo de trastornos linfoproliferativos en que se engloban todos los linfomas que no encajan dentro de la definición de linfoma de Hodgkin. Por tanto, son neoplasias linfoides que pueden presentar fenotipo de linfocitos B, T o de células NK (natural killer).
A grandes rasgos, en los LNH una célula linfoide inmadura, detenida en un determinado estadio madurativo, se reproduce de forma incontrolada, causando con el tiempo el aumento de tamaño del órgano en el que se producen. Dado que el tejido linfático se encuentra en todo el cuerpo, los linfomas pueden aparecer en cualquier parte del organismo y, a partir de allí, diseminarse a otros órganos y tejidos. En la mayoría de los casos empiezan con una infiltración en un ganglio linfático (formas ganglionares), pero algunos subtipos específicos pueden aparecer diseminados y estar restringidos a otros órganos como el aparato digestivo, la piel, el cerebro, el bazo, el riñón u otros órganos (formas extraganglionares).
La clasificación de los LNH de la OMS establece dos grandes grupos, según el origen: linfomas de células B (linfoma linfoblástico agudo de células B, leucemia de células pilosas, plasmocitoma, linfoma de Burkitt, etc.) y los linfomas de células T y de células NK (linfoma linfoblástico agudo precursor de células T, micosis fungoide, linfoma extranodal de células T, leucemia agresiva de células NK, linfoma anaplásico de células grandes, etc.). Los de linfocitos B representan el 80-90% de los LNH y los T el 10-20%, mientras que los de células citotóxicas NK tienen una frecuencia marginal. Tanto en las neoplasias linfoides B como en las T se distinguen dos tipos de transformación neoplásica, uno que se origina a partir de las células precursoras y el otro a partir de las células periféricas.
Desde un punto de vista clínico, los LNH se pueden dividir en dos grandes grupos en función de su velocidad
de crecimiento:
Los LNH representan el 4-5% de los nuevos casos de cáncer diagnosticados al año, ocupando el quinto lugar en frecuencia; así, por ejemplo, según el Instituto Nacional del Cáncer (NIH) estadounidense, en 2018 los LNH supusieron el 4,3% de los nuevos casos de cáncer en EE.UU. y el 3,3% de los fallecimientos por esta causa, situándose la tasa de supervivencia a 5 años entre 2008 y 2014 en el 71%. En las últimas décadas se ha registrado un aumento en las tasas de incidencia y de mortalidad por los LNH, principalmente en países industrializados; por ejemplo, se ha constatado un aumento especialmente acusado de las tasas de incidencia de los LNH en España e Italia. El aumento afecta a todos los grupos de edad adulta, aunque el mayor aumento se registra en los sectores de edad más avanzada de la población.
Si bien la mediana de presentación se sitúa en torno a los 50 años, los LNH pueden aparecer en cualquier edad de la vida, siendo algo más comunes entre los hombres y entre pacientes con enfermedades del sistema inmunitario (SIDA, inmunodeficiencias, receptores de trasplantes de órganos, enfermedades autoinmunes), infecciones (gastritis por Helicobacter, virus de Epstein-Barr), y pacientes tratados con quimioterapia o radioterapia. La incidencia general de LNH en España oscila entre los 10 y los 12 nuevos casos/100.000 habitantes/año. Aunque de forma menos frecuente, es un tipo de cáncer que también puede afectar a los niños, habiéndose descrito una incidencia para los LNH (no Burkitt) en nuestro país de 0,67 casos nuevos por 100.000 en niños de 0 a 14 años en el periodo 2000-2016; y de 0,54 casos nuevos por 100.000 para el linfoma de Burkitt en ese mismo periodo.
La etiopatogenia de los LNH varía entre los distintos tipos, aunque tienen en común ciertos factores de riesgo, como tener un sistema inmune debilitado (ya sea por una enfermedad hereditaria o tras un trasplante de órganos), edad elevada, antecedentes familiares, exposición a agentes tóxicos (herbicidas) e infecciones por algunos virus (virus linfotrópico de células T del ser humano tipo 1 –HTLV-1–, VIH o VEB) y bacterias (Helicobacter pylori).
De forma similar, Los síntomas de los LNH son muy variables y dependen de cada tipo específico de linfoma y de los órganos que estén afectados. Un gran porcentaje de pacientes son diagnosticados al detectarse una adenopatía. Resulta característico que los pacientes sintomáticos pueden presentar fiebre, sudoración nocturna, pérdida de peso (cuando estas tres manifestaciones se presentan a la vez se habla de grupo de síntomas B), fatiga e infecciones de repetición. También pueden producirse manifestaciones como consecuencia del crecimiento del tamaño del bazo (molestias abdominales), de la compresión de un órgano por un tumor de gran tamaño (tos, dolor lumbar o abdominal) o del mal funcionamiento de un órgano por su infiltración por las células cancerosas.
El tratamiento de los LNH es variable en función del tipo de linfoma, la edad y el estado general del paciente, la extensión de la enfermedad y la progresión de la misma. Como norma general, los linfomas indolentes, a pesar de que a veces pueden hallarse muy extendidos por el organismo, tienen una muy lenta evolución, por lo que pueden no requerir tratamiento inmediato tras el diagnóstico; en ellos, la respuesta al tratamiento suele ser buena pero casi nunca curativa del todo, por lo que, con el tiempo, el linfoma volverá a aparecer. Contraria y paradójicamente, los LNH agresivos, que tienen una evolución más rápida que obliga a tratarlos rápidamente, suelen responder muy bien al tratamiento, siendo frecuente alcanzar la remisión completa de la enfermedad, con ausencia de síntomas y de enfermedad y, en muchos casos, su curación.
El tratamiento de los linfomas indolentes o de bajo grado puede variar entre los distintos subtipos. Así, en el linfoma folicular (el más frecuente) y en pacientes asintomáticos, la abstención terapéutica se considera la actitud más adecuada; en pacientes con enfermedad localizada, la radioterapia puede ser el mejor tratamiento. Para el resto de pacientes, no existe un tratamiento que pueda considerarse estándar, pero los actuales se basan en el empleo de rituximab, un anticuerpo monoclonal contra el receptor CD20, asociado a uno o varios –hasta 4– agentes quimioterápicos clásicos, antineoplásicos del tipo de los agentes alquilantes (ciclofosfamida, bendamustina) o antimetabolitos (fludarabina). Por ejemplo, se consideran las siguientes combinaciones más comunes: CHOP+R (la asociación de rituximab a un régimen cuádruple de ciclofosfamida, doxorrubicina, vincristina y prednisona), CVP+R (rituximab más ciclofosfamida, vincristina y prednisona), FCM+R (rituximab más fludarabina, ciclofosfamida y mitoxantrona) o FND+R (rituximab más fludarabina, mitoxantrona y dexametasona).
Ese tipo de tratamientos permite alcanzar tasas de respuesta global de hasta un 90%, que son completas hasta en el 60% de los casos primarios. La duración media de la respuesta oscila entre 1 y 4 años. Si no hay respuesta al tratamiento o la enfermedad vuelve a aparecer, puede plantearse un esquema quimioterápico diferente, generalmente añadiendo un escalón más en combinación, que podrá incluir también otros anticuerpos monoclonales o fármacos contra nuevas dianas terapéuticas. En algunos casos, en función de la respuesta, la edad y estado general del paciente, se podrá considerar realizar un TPH de médula ósea autólogo o alogénico.
En ciertos tipos de linfomas, como los MALT, el tratamiento se basa en combatir el estímulo antigénico que ha originado la transformación neoplásica (infecciones, procesos inflamatorios locales, enfermedades autoinmunes, etc.) por lo que el tratamiento puede consistir únicamente en el tratamiento de la infección o la patología causal, si bien en estadios avanzados o si el linfoma no responde a estos tratamientos suele ser necesario añadir al mismo quimioterapia convencional, asociada o no a rituximab.
Por otra parte, el tratamiento de los linfomas agresivos o de alto grado también es variable en función del tipo de linfoma, su extensión, la edad y estado general del paciente. Así, en el más frecuente, el linfoma difuso de células grandes B, el esquema quimioterápico más empleado es el CHOP+R, con una frecuencia de administración y número de ciclos variable según cada caso. La radioterapia puede ser efectiva para tratar áreas afectas localizadas o masas muy grandes. El TPH (normalmente autólogo y, más excepcionalmente, alogénico) queda limitado en estos LNH agresivos a los pacientes refractarios al tratamiento de primera línea o en recaída, siempre que la enfermedad permanezca quimiosensible.
En líneas generales, la radioterapia clásica se utiliza solo en el tratamiento de LNH para reducir adenopatías que causan síntomas compresivos en pacientes refractarios a otros tratamientos. Aunque la irradiación corporal total en pequeñas dosis ha arrojado resultados satisfactorios en algunas ocasiones, el uso más habitual es en administración local en zonas de adenopatías, hígado o bazo para obtener una paliación sintomática transitoria.
Incluso con un tratamiento óptimo, el pronóstico es muy variable para cada subtipo de LNH. Así, por ejemplo, en los linfomas foliculares, a pesar de la escasa probabilidad de curación completa con el tratamiento, la supervivencia es muy prolongada y la mayoría de pacientes viven más de 10 años después del diagnóstico. Los linfomas agresivos, como ya se ha indicado, suelen responder muy bien al tratamiento y la probabilidad de curación de un paciente con un linfoma difuso de células grandes que pueda recibir el tratamiento de forma correcta se sitúa en el 60-80%. El Registro RETI-SEHOP recoge unas cifras supervivencia a 5 años del 89% para los pacientes con LNH –incluyendo linfomas de Burkitt– diagnosticados en España en la cohorte de 2009-2011.
Tras aportar una visión general de los fármacos específicos más interesantes usados en el tratamiento del LNH (aunque hay más), se abordarán las particularidades de algunos LNH que con más frecuencia afectan específicamente a niños.
La bendamustina (Levact® y EFG) es un agente antitumoral alquilante cuyos efectos antineoplásicos y citocidas se basan esencialmente en un entrecruzamiento de las cadenas del ADN dobles y simples por alquilación, alterando, como resultado, las funciones de la matriz del ADN y de su síntesis y reparación. Ha demostrado una clara superioridad sobre el clorambucilo en pacientes con leucemia linfocítica crónica o linfoma linfocítico de células pequeñas (LLCP)10 intolerantes a la fludarabina, consideraba como tratamiento de referencia (en combinación con rituximab y ciclfosfamida), si bien la toxicidad de bendamustina también es algo mayor que la del clorambucilo; con todo, el balance beneficio/riesgo parece claramente favorable a la bendamustina. También se ha mostrado eficaz en pacientes con LNH indolentes que hayan progresado durante o en los 6 meses siguientes a un tratamiento con rituximab o un régimen que contenga rituximab.
Los análogos de purinas, fludarabina y cladribina, son algo más eficaces y menos tóxicos que los agentes alquilantes, produciendo remisiones más prolongadas en LLCP, si bien tampoco han demostrado ventajas sustanciales con respecto a la supervivencia global, al menos en monoterapia. En pacientes previamente no tratados, con fludarabina se obtiene un 30-40% de respuestas completas y un 8-20 % de respuestas parciales, con una mediana de duración de la respuesta de más de 2 años y una supervivencia estimada a los 6 años del 80%. La respuesta máxima se obtiene en 2-3 meses y, en caso de no conseguirse respuesta, se debe detener el tratamiento. Los pacientes con enfermedad refractaria o en recaída tras diversos tratamientos previos han demostrado respuestas importantes en el 30-60% de los casos (15-40% respuestas completas y, adicionalmente, un 15-50% respuestas parciales). Por su parte, la cladribina actúa tanto sobre linfocitos (proliferantes o en reposo) como monocitos. En pacientes no tratados previamente se ha obtenido un 25% de respuestas completas y un 60% de respuestas parciales, con una mediana de duración de la respuesta superior a 8 meses. En pacientes previamente tratados, se ha obtenido un 5% de respuestas completas y un 40% de respuestas parciales, con una mediana de duración de la respuesta de 4 meses.
La pixantrona (Pixuvri®) es una aza-antraquinona citotóxica del grupo de los antineoplásicos intercalantes de ADN, que se diferencia de las antraciclinas (doxorrubicina y otras) y antraquinonas (mitoxantrona) autorizadas en que es solo un inhibidor débil de la topoisomerasa II11, y en que alquila directamente el ADN: se intercala entre las parejas de bases de la doble hebra y forma aductos estables de ADN mediante la formación de enlaces covalentes, con la consiguiente estabilización del complejo ADN-topoisomerasa II y deformación de la estructura normal, todo lo cual provoca roturas de la cadena doble y el bloqueo de la síntesis de nuevo ADN. Además, como incorpora un heteroátomo de nitrógeno en la estructura anular y no tiene grupos cetónicos, tiene un menor potencial para la generación de especies reactivas del oxígeno, fijación de hierro y formación de metabolitos alcohólicos que se consideran responsables de la toxicidad cardiaca de las antraciclinas. El medicamento fue autorizado para la monoterapia en pacientes adultos con LNH de linfocitos B agresivos, multirrecidivantes o resistentes al tratamiento.
Por otro lado, los receptores CD20 son antígenos transmembrana que están presentes en la superficie celular de los linfocitos B, tanto normales como malignos (incluyendo las formas maduras, proliferantes y diferenciadas), donde actúan como receptores moleculares del antígeno Bp35, una proteína fosforilada responsable de la restricción de la diferenciación de los linfocitos B, que es expresada durante las fases más precoces. Esta circunstancia condujo al desarrollo de anticuerpos específicos, como rituximab (Mabthera® y los biosimilares Rixathon®, Truxima®, Riximyo® y Ruxience®), capaces de provocar la destrucción selectiva de linfocitos B –aunque sin distinción entre fisiológicos y malignos– como consecuencia de una combinación, en diferentes grados, de efectos celulares de citotoxicidad y fagocitosis mediadas por anticuerpos, así como apoptosis independiente de la cascada de caspasas (inducción directa) y citotoxicidad dependiente del complemento. Se considera que la citotoxicidad y la fagocitosis celulares mediadas por anticuerpos son los mecanismos antineoplásicos principales de este tipo de anticuerpos anti-CD20.
El rituximab ha demostrado eficacia en pacientes con leucemia linfocítica crónica intensamente pretratados (50% de respuesta global), con posibilidad de retratamiento (44% de respuesta global) y disminución del tamaño del tumor (87% de los pacientes). En combinación con quimioterapia CHOP (ciclofosfamida, doxorubicina, vincristina, prednisolona), la tasa de respuesta se eleva hasta un 95% de los pacientes. La combinación de rituximab con fludarabina y ciclofosfamida requiere una función renal normal y un estado general adecuado, además de que la presencia de determinadas anomalías citogenéticas (como la del17p) son predictores de mala respuesta al tratamiento, lo que limita su utilidad. Cuando la fludarabina no sea apropiada, se considerará el clorambucilo, o mejor aún la bendamustina, como alternativas para la primera línea de tratamiento, especialmente en pacientes de edad avanzada y comorbilidad importante.
Actualmente, rituximab tiene aprobada una única indicación en pacientes pediátricos: en combinación con quimioterapia, para el tratamiento de pacientes de edad ≥ 6 meses hasta < 18 años con linfoma B difuso de célula grande (LBDCG) CD20 positivo en estado avanzado no tratado previamente, linfoma de Burkitt/leucemia de Burkitt (leucemia aguda de células B maduras) o linfoma similar a Burkitt. Además, tiene diversas indicaciones en pacientes adultos: i) en combinación con quimioterapia en el tratamiento de pacientes adultos con LNH folicular estadio III-IV que no hayan sido tratados previamente, sean quimiorresistentes o estén en su segunda o posterior recidiva tras la quimioterapia; ii) en el tratamiento de mantenimiento en pacientes adultos con linfoma folicular que hayan respondido al tratamiento de inducción; y iii) en combinación con quimioterapia CHOP en el tratamiento de pacientes adultos con LNH difuso de células B grandes CD20+.
Continuando con los agentes biológicos, el obinutuzumab (Gazyvaro®, autorizado en 2014) es un anticuerpo monoclonal humanizado de tipo IgG1κ modificado por glicoingeniería que también se une y actúa específicamente sobre el bucle extracelular de CD20 en la superficie de linfocitos pre-B y B maduros malignos y no malignos, pero no en células madre hematopoyéticas, células pro-B, células de plasma normales u otro tejido normal. La modificación de la parte Fc de la molécula aumenta la afinidad por los receptores de FcɣRIII en células efectoras inmunes, tales como células NK, macrófagos y monocitos, en comparación con los anticuerpos no modificados.
Obinutuzumab forma parte del tipo II de anticuerpos anti-CD20 utilizados en terapéutica. Mientras que los de tipo I –como rituximab– actúan fundamentalmente mediante un potente efecto citotóxico dependiente del complemento, mediado por la inducción de translocación del CD20 en grandes microdominios lipídicos, los de tipo II –como obinutuzumab– tienen un efecto citotóxico directo (muerte celular directa, citotoxicidad celular dependiente de anticuerpos y fagocitosis celular dependiente de anticuerpos por el reclutamiento de células efectoras inmunes FcɣRIII+) mucho más marcado que su efecto dependiente del complemento. Así, los de tipo II son significativamente más potentes que los de tipo I en la inducción de apoptosis de células CD20+, tanto normales como malignas, permitiendo alcanzar una mayor depleción de linfocitos B que el rituximab.
En su caso, obinutuzumab está autorizado para el tratamiento, en combinación con clorambucilo, de pacientes adultos con leucemia linfática crónica (LLC) no tratados previamente y con comorbilidades que les hace no ser adecuados para el tratamiento basado en una dosis completa de fludarabina. Adicionalmente, se ha aprobado frente al LNH folicular: en pacientes no tratados previamente (naïve) en combinación con quimioterapia seguido de monoterapia de mantenimiento en pacientes que alcanzan algún tipo de respuesta, y, en combinación con bendamustina seguido de monoterapia en mantenimiento, en pacientes que no han respondido o han progresado en ≤ 6 meses después del tratamiento con un régimen con rituximab. Sin embargo, no se ha establecido su seguridad y eficacia en niños y adolescentes de < 18 años de edad, por lo que no se ha autorizado en población pediátrica.
El ibritumomab tiuxetan (Zevalin®) es un anticuerpo monoclonal que une de forma específica al receptor CD20 presente en la gran mayoría de los linfocitos B humanos y, estando ligado a un agente quelante (el tiuxetano), actúa como anclaje de un radioisótopo del itrio (90Y), un radionúclido emisor de radiación beta de baja penetración (5-10 mm) y con una vida media de 64 h. Se trata, por consiguiente, de un agente quimioinmunoradioterapéutico, en el que la fracción biológica es un anticuerpo IgG1 anti-CD20, que “localizará” a los linfocitos B, tanto fisiológicos como malignos, que es la población celular cuya malignificación conduce al desarrollo del LNH folicular, y una vez unido a ellos, permite que el 90Y ejerza su capacidad de destruir las células diana y las células vecinas. El tratamiento previo con rituximab –a una dosis más reducida en comparación con la monoterapia autorizada– es necesario para eliminar las células B circulantes, lo que permite que el ibritumomab tiuxetan marcado con 90Y libere la radiación a los linfocitos B del linfoma de un modo más específico.
Esta forma de inmunoradioterapia para las fases avanzadas de LNH folicular da lugar a tasas relativamente altas de respuesta, superiores al rituximab, incluso en pacientes refractarios a otros tratamientos antineoplásicos (incluyendo al propio rituximab), con respuestas que llegan a alcanzar en una minoría de pacientes una duración superior a 3 años, aunque dada la fase avanzada de desarrollo del linfoma y la refractariedad a otras terapias, previsiblemente no puede considerarse éste como un parámetro de especial relevancia clínica. El medicamento, una vez marcado con 90Y, está indicado en el tratamiento de pacientes adultos con LNH folicular de células B CD20+ en recaída o refractario a rituximab, si bien no se ha establecido el beneficio después de rituximab asociado a la quimioterapia; también como tratamiento de consolidación después de la inducción de la remisión en pacientes con LNH folicular no tratados anteriormente.
Por último, se puede destacar la incorporación reciente de un agente quimioterapéutico específico como el idelalisib (Zydelig®, comercializado en España en 2016), que inhibe selectivamente la unión del ATP al dominio catalítico, y con ello, la actividad12 de la fosfatidilinositol 3-cinasa p110δ (PI3Kδ), cuya actividad enzimática está potenciada en las neoplasias malignas de linfocitos B, al estar implicada en numerosas vías bioquímicas de señalización que impulsan la proliferación, supervivencia, migración y retención de las células tumorales en los tejidos linfoides y en la médula ósea. El efecto final del proceso es la inducción de la apoptosis y la inhibición de la proliferación de las células tumorales primarias y de las líneas celulares derivadas de los linfocitos B tumorales. El medicamento ha sido autorizado, en combinación con rituximab, para el tratamiento de pacientes adultos con leucemia linfocítica crónica que han recibido ≥ 1 tratamiento anterior, o bien como tratamiento de 1ª línea en presencia de deleción en 17p o mutación de TP53 en pacientes que no son adecuados para recibir ningún otro tratamiento. Además, se indica en monoterapia para el tratamiento de los pacientes adultos con linfoma folicular refractario a 2 líneas de tratamiento anteriores.
Las peculiaridades del tratamiento en los distintos subtipos de LNH fueron ampliamente revisados y publicados en PAM (Cuéllar, 2018).
El linfoma de Burkitt, también conocido como linfoma de células pequeñas no hendidas, es un LNH agresivo de células B poco frecuente y que afecta sobre todo a pacientes jóvenes, siendo el subtipo de LNH mayoritario en población pediátrica. En un amplio porcentaje de casos se puede apreciar una translocación entre alguno de los cromosomas de los pares 8 y 14 –t(8;14)–, que se traduce en la activación del protooncogén c-MYC. Se distinguen 3 formas clínicas con características y manifestaciones distintas:
El tratamiento de primera línea debe iniciarse de inmediato tras el diagnóstico y se basa en la quimioterapia intensiva con dosis elevadas. El tratamiento más habitual asocia ciclofosfamida, metotrexato, citarabina, rituximab y la profilaxis de la infiltración del sistema nervioso central. El TPH no está indicado como tratamiento de primera línea y se reserva para pacientes con mal pronóstico.
El linfoma anaplásico de células grandes (LACG) es una variedad de LNH muy agresiva que deriva de linfocitos T y que debe separarse del resto de los linfomas de células T por su pronóstico más favorable y su respuesta al tratamiento. Se diferencian dos formas, la sistémica y la cutánea (esta última es común entre pacientes infectados por el VIH y se suele presentar como un nódulo benigno que desaparece por sí solo o se disemina a muchos lugares en la piel aunque sin carácter canceroso), y en conjunto representan aproximadamente el 3% de todos los LNH en adultos y el 10-30% de los linfomas infantiles.
Tiene la peculiaridad de expresar la proteína CD30 y la evolución clínica depende en buena medida del estatus ALK: aunque el LACG puede presentarse a cualquier edad, el subtipo ALK+ es más frecuente en menores de 30 años y predomina en varones, mientras que la forma ALK- es más frecuente en mayores, sin diferencia entre sexos. La cinasa del linfoma anaplásico (ALK, por sus siglas en inglés), también conocida como receptor de la tirosina cinasa de ALK o CD246, se trata de un enzima que en los seres humanos está codificada por el gen ALK y que desempeña un papel importante en el desarrollo del cerebro y ejerce sus efectos sobre neuronas específicas. Sin embargo, el gen ALK puede actuar como un oncogén de tres maneras diferentes: a través de la formación de un gen de fusión con otros genes, mediante la obtención de copias adicionales de genes o con mutaciones del código de ADN para el propio gen. Más de la mitad de los casos de LACG se asocian con una determinada translocación entre los cromosomas 2 y 5, denominada t(2;5)(p23;q35), que crea un gen de fusión de ALK y de la nucleofosmina (NPM), de tal manera que la fracción 3’ de la ALK (proveniente del cromosoma 2 y que codifica para el dominio catalítico), está fusionada a la porción 5′ de NPM, proveniente del cromosoma 5. El producto de fusión formado (NPM/ALK) se traduce en una proteína quimérica denominada p80, que es oncogénica y característica de este tipo tumoral.
Al momento del diagnóstico, la mayoría de los pacientes con LACG presentan enfermedad avanzada, con síntomas sistémicos (sobre todo, fiebre elevada) y afectación extranodal. La médula ósea suele estar infiltrada en un elevado número de casos. En la mayoría de los pacientes el pronóstico es favorable, con una supervivencia media a los 5 años de aproximadamente el 60%. Se considera factor de buen pronóstico la regresión espontánea de las lesiones y de mal pronóstico la extensión extracutánea del tumor.
En cuanto al tratamiento, la combinación de quimioterapia de tipo CHOP o ABVD, a la que en estadios localizados puede añadirse la radioterapia, constituye la base terapéutica del LACG, que permite alcanzar una remisión completa en el 70-80% de los pacientes ALK+. Más recientemente, varios agentes nuevos han ofrecido la oportunidad de un cambio en el paradigma para el manejo de las formas de LACG, tanto quimiosensibles como resistentes a la quimioterapia. El desarrollo de inhibidores de ALK después de la identificación del gen de fusión EML4-ALK en el cáncer de pulmón no microcítico ha abierto nuevas posibilidades para el LACG ALK+. La expresión uniforme de CD30 en la superficie celular de LACG ha dado la oportunidad para la terapia con anticuerpos anti-CD30. Por otro lado, la reevaluación de la vinblastina, que ha demostrado una notable actividad como agente único incluso frente a una enfermedad recurrente, ha llevado a la consideración de un enfoque revisado para la terapia de primera línea. El advenimiento de inmunoterapias, como los anti-PD-1 nivolumab y pembrolizumab también ha proporcionado otra opción (Prokoph et al., 2018).
El linfoma linfoblástico es un tipo de LNH agresivo que afecta especialmente a niños. Se clasifica según si es de precursores B o T, siendo la afectación de células T lo más habitual. Las células tumorales son las mismas que las de la leucemia linfoblástica aguda (LAL); de hecho, la enfermedad solo se clasifica como LNH linfoblástico y se trata como tal si la proporción de blastos en la médula ósea es inferior al 25% y no existen blastos en sangre periférica; en caso contrario, se clasifica y trata como leucemia linfoblástica aguda (LAL). En el momento del diagnóstico, más del 90% de los pacientes se halla en estadios avanzados (III y IV) de la enfermedad y un 60% acaba desarrollando LAL.
Desde el punto de vista terapéutico, la combinación de quimioterapia habitual en otros linfomas agresivos, como el régimen CHOP, no ofrece buenos resultados. Por ello, el tratamiento debe basarse en esquemas propios de la LAL –anteriormente comentados– para conseguir tasas de remisión elevadas (70-90%) en niños, que serán mucho menores en adultos. Debido a la pobre respuesta al tratamiento en adultos, en ocasiones se recomienda la administración de altas dosis de quimioterapia seguidas de un TPH autólogo o, fundamentalmente, alogénico.
Por último, merece una mención el linfoma difuso de células grandes B (LDCGB), que se comenta aquí no porque afecte a niños primordialmente, sino por los avances recientes en su tratamiento. Se trata de un LNH agresivo, con una gran prevalencia, que representa alrededor del 30% de todos los LNH, y del cual se ha reportado una incidencia global de 50-60 nuevos casos por millón de habitantes y año, creciente a medida que aumenta la edad media de la población, ya que, aunque se observa en cualquier edad, la mediana de los pacientes con LDCGB está en torno a 55 años.
La evolución de este tipo de linfoma es agresiva y su pronóstico depende mucho de la edad del paciente, su estado general, la extensión del tumor y la respuesta al tratamiento. Se han descrito dos subtipos: el linfoma difuso de tipo centro germinal y el de célula B activada; este último tiene peor pronóstico. No obstante, hasta el 80% de los pacientes jóvenes se pueden curar, disminuyendo esta probabilidad con el aumento de la edad.
El tratamiento de primera línea de estos linfomas se basa en la asociación de quimioterapia y radioterapia sobre áreas afectas localizadas o de gran tamaño (masa mediastínica). El esquema quimioterápico más empleado en la actualidad es el CHOP+R, con una frecuencia de administración y un número de ciclos variable según cada caso. La radioterapia puede ser efectiva para tratar áreas afectas localizadas.
Para el tratamiento de pacientes con LDCGB recidivante/refractario, el tratamiento suele consistir en quimioterapia de rescate basada en platino, con la posible adición de rituximab. El TPH autólogo es utilizado posteriormente en menos de la mitad de pacientes, refractarios al tratamiento de primera línea o en recaída, en que la enfermedad permanezca quimiosensible. En los no candidatos al TPH (> 50%) o aquellos que recidivan tras el mismo, se han descrito tasas de supervivencia libre de eventos a los 4 años del 46-56%, y solo pueden tratarse con pixantrona en monoterapia, que aporta aumentos de supervivencia libre de eventos y supervivencia global, respectivamente, de 2,7 y 2,6 meses (vs. quimioterapia clásica). Pero, tras 2 o más líneas, no hay tratamiento estándar y la calidad de vida es muy pobre. De manera interesante, es precisamente en ese escenario clínico, solo en pacientes adultos, en el que se han autorizado, con resultados clínicos realmente prometedores, los dos primeros medicamentos CAR-T anti-CD-19 de fabricación industrial comercializados en España: tisagenlecleucel (Kymriah®) –comentado en el apartado de tratamiento de la leucemia linfoblástica aguda–, y axicabtagén ciloleucel (Yescarta®), de fundamento y mecanismo prácticamente idéntico al primero (Fernández Moriano, 2019e).
Como se ha sugerido anteriormente, el amplio desconocimiento de la etiología y de los factores de riesgo implicados en la patogenia de los cánceres infantiles imposibilita una actuación eficaz sobre una prevención primaria. Para ello, es condición sine qua non avanzar en el conocimiento de la etiopatogenia de los cánceres pediátricos y resulta necesario iniciar proyectos de investigación que evalúen exhaustivamente la participación de los factores de riesgo constitucionales y medioambientales presentes en esa población pediátrica, a fin de que algunos de ellos puedan prevenirse.
Hasta entonces, el núcleo central de la lucha contra esta enfermedad reside en un diagnóstico adecuado y en estadios tempranos13, crucial para el pronóstico, y en la optimización del tratamiento farmacológico y/o quirúrgico, que están enfocados a una prevención secundaria que busca disminuir la progresión a fases avanzadas de las neoplasias malignas, con terapias menos agresivas y mejor calidad de vida de los pacientes, y una reducción del riesgo de complicaciones y mortalidad. A este respecto, uno de los mejores ejemplos entre los cánceres pediátricos es el cribado del neuroblastoma en las primeras fases de la vida mediante la detección precoz de los metabolitos de las catecolaminas.
Se debe mencionar también la necesidad de que padres y profesionales sanitarios se familiaricen y sean capaces de detectar precozmente los signos de alarma sugerentes de leucemias o linfomas en la edad pediátrica, que se citarán en el apartado siguiente. De igual modo, es preciso advertir que la emergencia sanitaria provocada por la COVID-19 a nivel mundial ha complicado aún más el ya de por sí difícil proceso diagnóstico y terapéutico de los tumores por el retraso de las revisiones médicas. Esto ha supuesto que algunas voces de expertos se alcen ante el diagnóstico de algunos tumores en estadios avanzados que podrían haberse detectado en revisiones médicas rutinarias. Resulta necesario concienciar a la población, por tanto, de que las reticencias a acudir a un centro médico a una revisión o ante la aparición de cierto signo/síntoma clínico por el miedo al contagio por SARS-CoV-2 pueden conllevar un mayor riesgo de desarrollar una enfermedad potencialmente más grave que la propia COVID-19.
Por otra parte, también se debe citar en este apartado la relevancia de la prevención de otras patologías en pacientes pediátricos oncológicos. Por ejemplo, estos pacientes requieren la adaptación de los calendarios vacunales, ya que presentan necesidades especiales de vacunación una vez finalizado el tratamiento, sobre todo, de quimioterapia. Un reciente estudio observacional retrospectivo (Fernández-Prada et al., 2018) evaluó los datos vacunales –de acuerdo a las indicaciones oficiales del Comité Asesor de Vacunas de la Asociación Española de Pediatría– de una cohorte de 99 niños de 0 a 14 años que recibieron quimioterapia en un hospital entre 2009 y 2015. Se incluyeron en el análisis los datos de 51 pacientes (71% varones, 45% con tumores hematológicos), de los cuales el 71% tenía registrada la recepción de alguna vacuna tras el tratamiento, sin hallarse diferencias estadísticamente significativas entre el cumplimiento del calendario y el tipo de tumor, el sexo o la edad. En su mayoría, los pacientes habían recibido la vacuna difteria-tétanos-tosferina o difteria-tétanos (55%), meningococo C (41%) y gripe estacional (39%), siendo la tasa de adaptación de calendario posquimioterapia de solo el 10%. Así, la vacuna frente a neumococo conjugada 7valente o 13valente fue administrada en el 22% de los niños evaluados, pero solo se completó con polisacarídica 23valente en el 18% de los casos; ninguno recibió vacunación frente a hepatitis A. Los autores concluyen que existe un importante margen de mejora en la adaptación de la vacunación posquimioterapia en niños con cáncer. Frente a ello, la participación de los profesionales sanitarios en programas de formación y la derivación de estos pacientes a las específicas Unidades de Vacunas podría mejorar la tasa de adaptación garantizando una correcta inmunización.
Todos los profesionales farmacéuticos, desde sus diversos ámbitos de actuación y competencias, pueden contribuir al adecuado asesoramiento y asistencia sanitaria a los pacientes pediátricos con cánceres hematológicos y a sus familias. Teniendo en cuenta las particularidades comentadas en la presente revisión, lo más habitual es que la administración parenteral de los ciclos de quimioterapia antineoplásica y de los tratamientos más recientes de inmunoterapia se hagan en el medio hospitalario –para monitorizar directamente la respuesta del paciente y atender de forma inmediata cualquier complicación–, contexto en el que la figura del farmacéutico especialista cobra una especial relevancia en la consecución de los objetivos del proceso farmacoterapéutico. Una reseña merece su implicación en los tratamientos con las innovadoras terapias CAR-T, como se verá más adelante.
No obstante, la gran mayoría de niños con cáncer retorna a continuación a sus domicilios y, en muchos casos, vuelven a realizar su vida cotidiana, si bien continuando con un tratamiento antitumoral por periodos prolongados; además, cada vez son más los tratamientos –de mantenimiento– que se administran por vía oral en el ámbito ambulatorio. En esos casos, en que la adherencia representa un pilar fundamental del tratamiento, el papel del farmacéutico comunitario es también crucial para alcanzar el éxito terapéutico.
Así, atendiendo al hecho de que cada día más dos millones de pacientes y usuarios acuden a las más de 22.000 farmacias comunitarias españolas, y que en ellas se ofrecen al año más de 182 millones de consejos sanitarios, parece evidente el potencial divulgador del farmacéutico como profesional sanitario, así como su incuestionable papel para canalizar hacia el médico a personas con problemas relevantes de salud, para un estudio clínico detallado. La oficina de farmacia constituye un centro accesible y ubicuo capaz de suministrar una información rigurosa, pieza clave para aumentar la visibilidad de las neoplasias hematológicas en la sociedad, y ofrecer un servicio sanitario de máximas garantías y con la debida confidencialidad. Contribuye también a la detección precoz de este tipo de tumores, a la promoción de un uso racional de los medicamentos (a fin de prevenir los problemas relacionados con los mismos), y facilita la disponibilidad de numerosos medicamentos oncológicos, con claras implicaciones en la sostenibilidad del Sistema Nacional de Salud.
Dado el alto impacto sanitario y emocional que tienen estas patologías, con la integración efectiva del farmacéutico en los equipos multidisciplinares de atención primaria y especializada, se pueden identificar varias vías asistenciales enfocadas al abordaje de los pacientes y al asesoramiento práctico a familiares.
El farmacéutico puede y debe aportar a los pacientes información fácilmente comprensible, pero con rigor científico, sobre: a) los medicamentos: incidiendo sobre su objetivo y mecanismo, las peculiaridades de conservación (si las hubiera), el momento óptimo de administración, la posibilidad e importancia de interacciones con otros medicamentos (incluidos los de automedicación), etc.; y b) su pauta de administración: se puede aconsejar la adaptación de la toma coincidiendo con eventos cotidianos o aportar diagramas que ayuden a relacionar la medicación con hábitos de vida.
A la hora de afrontar una enfermedad como una neoplasia pediátrica, es importante transmitir, con la sensibilidad adecuada, que se trata de un proceso grave pero curable en un alto porcentaje de pacientes. No obstante, los tratamientos quimioterapéuticos son comúnmente largos y tienen una toxicidad para nada desdeñable que interferirá de forma importante con la calidad de vida de los pacientes. En ese contexto, la vida de toda la familia deberá reorganizarse, para lo cual el trabajo en las Unidades de Oncopediatría hospitalarias de psicólogos, trabajadores sociales y voluntarios se antoja crucial. Se debe advertir a los familiares de que, aun con un tratamiento exitoso, los niños convivirán a largo plazo con un riesgo notable de efectos adversos y recaídas, y que estarán sometidos “de por vida” a revisiones médicas frecuentes, debiendo vigilar más de cerca su estado de salud y el cumplimiento de hábitos de vida saludables.
En esa línea, uno de los aspectos más importantes para asegurar una mejor salud es la educación nutricional. Por un lado, el propio cáncer y sus tratamientos pueden desencadenar un estado de malnutrición debido a una disminución de la ingesta por afecciones del tubo digestivo (que, en el ámbito hospitalario muchas veces hacen necesaria la alimentación parenteral, con sonda nasogástrica o a través de gastrostomía) y/o a una situación de anorexia característica de la fisiopatología de algunas leucemias o linfomas. Las alteraciones nutricionales se suelen asociar también con el tipo o estadio del tumor, ya que los más avanzados cursan con una situación de hipermetabolismo debida a la respuesta inmunitaria e inflamatoria ante la enfermedad. E, incluso, una alimentación inadecuada puede aumentar la incidencia de determinados tipos de cánceres en la edad adulta.
Para una mayor información se puede recurrir a publicaciones específicas que recogen las recomendaciones dietéticas más beneficiosas para pacientes oncológicos pediátricos (Beas et al., 2016; Aguerri, 2019). Entre ellas, se pueden destacar: i) presentar los alimentos de forma atractiva y no ofrecer alimentos que resulten desagradables; ii) aumentar el número de comidas, disminuyendo la cantidad de alimentos en cada una de ellas; y iii) proporcionar preferentemente alimentos de alto contenido calórico y/o proteico: carnes, pescados, huevos, legumbres, cereales, derivados. Se debe tener en cuenta que, además de impedir una adecuada tolerancia al tratamiento, la malnutrición agrava el estado de inmunosupresión al que está sometido el paciente oncológico, debido al menos en parte a la disminución de las proteínas circulantes, del metabolismo oxidativo y del filtrado glomerular, lo cual provocará una reducción de la vida media de los fármacos y, por tanto, de su acción, así como un aumento de toxicidad. Otro elemento a considerar es que se puede alcanzar cierto grado de prevención primaria asegurando un consumo elevado de frutas y verduras, así como de suplementos de ácido fólico, durante la gestación; se ha constatado cierto efecto protector de esta suplementación contra la leucemia, el neuroblastoma, y otros tumores del SNC en niños (Bhoite, 2016).
Por otra parte, conviene recordar a pacientes y familiares que los cánceres hematológicos y su tratamiento implican un riesgo incrementado de padecer infecciones en comparación a un sujeto sano. Las infecciones de las vías respiratorias son frecuentes y, potencialmente, las más graves, por lo que se pueden recomendar ciertas medidas básicas para reducir el número de episodios: evitar el contacto directo con familiares o personas sintomáticas, extremar la higiene o evitar en la medida de lo posible las aglomeraciones en épocas de gripe. Tal y como se está repitiendo en la pandemia por COVID-19, el lavado frecuente de manos también adquiere relevancia en determinadas situaciones (por ejemplo, en el interior de hospitales), y en momentos de especial susceptibilidad (por ejemplo, tras un trasplante) puede ser recomendable portar una mascarilla quirúrgica o filtrante que evite la transmisión aérea de microorganismos patógenos. Otras medidas sencillas para minimizar la probabilidad de infecciones son: una dieta variada y equilibrada, una buena hidratación, ejercicio físico regular y moderado y evitar el tabaco o las aglomeraciones en ambientes cerrados.
Además, desde la farmacia se puede ejercer una acción social crucial, mediante el asesoramiento y orientación de los niños y adolescentes con nuevo diagnóstico, y, sobre todo, de sus padres (y hermanos), para que se enrolen en asociaciones de pacientes que les brindarán información muy diversa, acompañamiento, apoyo (incluyendo el aspecto económico) y diversos servicios socio-sanitarios, como asistencia psicológica. Dos buenas opciones son, por ejemplo, el foro de pacientes y ex-pacientes de la Fundación Josep Carreras o la Federación Española de Padres de Niños con Cáncer. Esta última es una entidad sin ánimo de lucro, constituida en 1990 y que coordina la actividad de 21 asociaciones integrantes repartidas por toda España, todas fundadas y presididas por madres y/o padres voluntarios afectados; su objetivo básico es el de fomentar y desarrollar las medidas necesarias para conseguir que los niños y adolescentes con cáncer tengan las máximas oportunidades de diagnóstico, tratamiento y curación.
Habida cuenta de que prácticamente ningún caso de cáncer pediátrico está relacionado con factores de riesgo evitables (a diferencia de otros tipos de cáncer en adultos, como el de piel o el de pulmón), se considera que en la actualidad la mejor medida preventiva para evitar las complicaciones y la mortalidad es la detección precoz, que pueda permitir un tratamiento temprano (ya que el momento de inicio es un factor decisivo en su eficacia). No obstante, ésta no es sencilla en el caso de tumores hematológicos, puesto que las manifestaciones no son específicas y se pueden confundir con otras afecciones, y a menudo no debutan clínicamente hasta fases avanzadas.
En este sentido, desde la farmacia comunitaria, el farmacéutico puede actuar como agente centinela teniendo presentes los signos de alarma que pueden sugerir o hacer sospechar de la presencia de una leucemia o un linfoma. Entre ellos, se pueden destacar los siguientes:
Para más detalles, se recomienda consultar la Guía de Atención Temprana de Cáncer en Niños y Adolescentes de la Sociedad Española de Hematología y Oncología Pediátricas (SEHOP). En cualquier caso, ante cualquiera de dichas manifestaciones, la actuación del farmacéutico debe incluir indispensablemente la derivación del paciente al pediatra para un diagnóstico preciso. Los niños con antecedentes en la familia pueden ser más propensos a desarrollar algunos tipos de tumores hematológicos con implicación de factores genéticos y se recomienda su monitorización más estrecha. También existe asociación entre algunos síndromes y el cáncer pediátrico, como entre el síndrome de Down y la leucemia, o entre la neurofibromatosis y los tumores del sistema nervioso central.
Una vez establecido el diagnóstico de leucemia o linfoma, el farmacéutico, como profesional sanitario experto en el medicamento, debe velar por el uso seguro y eficaz de los mismos, para que los pacientes alcancen el máximo beneficio clínico. Esto es aplicable tanto en el entorno hospitalario, donde acudirán los pacientes a recibir los ciclos de quimioterapia o a la realización de un trasplante de progenitores hematopoyéticos, como, con igual o mayor relevancia, a nivel ambulatorio, pues hay que recordar que el farmacéutico comunitario conoce toda la medicación que utilizan estos pacientes, no solo la medicación prescrita frente al cáncer, sino también los tratamientos para enfermedades concomitantes, medicamentos que no necesitan prescripción, el uso de complementos alimenticios, etc. De forma general, las instrucciones dadas por parte del hematólogo/oncólogo tienen que ser estrictamente seguidas por el paciente, aunque pueden ir complementadas con otras del médico de atención primaria relativas a los cuidados y precauciones cotidianas o a la prescripción de cualquier tratamiento de continuación o complementario.
En el momento de la dispensación de cualquier medicamento prescrito en el curso de la enfermedad, el farmacéutico comprobará que el paciente cuente con toda la información necesaria para que el uso del mismo sea efectivo y seguro. Para ello, es conveniente averiguar si existe algún criterio que impida la dispensación, por ejemplo, alergia a algún componente del medicamento, una contraindicación absoluta o interacciones con otros medicamentos (o alimentos), una duplicidad o una situación fisiológica especial. Si es la primera vez que el paciente va a utilizar dicho medicamento, la labor del farmacéutico será asegurar que el paciente y los familiares conocen para qué es y cuál es su correcto proceso de uso. Si no fuera la primera vez (dispensación de continuación), evaluará si el medicamento está siendo eficaz y seguro, fundamentalmente verificando si ha habido cambios en el tratamiento (dosis, pauta posológica, duración del tratamiento, adición de nuevos medicamentos, etc.) y si el paciente ha experimentado algún problema que pudiera hacer sospechar de una posible reacción adversa, interacción, contraindicación, etc. Además de los medicamentos prescritos, en muchas ocasiones el propio paciente solicitará consejo al farmacéutico sobre los diferentes síntomas que van apareciendo.
Como en otras enfermedades que requieren tratamientos prolongados, incluyendo los episodios de recaída, la adherencia terapéutica ha sido descrita como uno de los factores de mayor influencia sobre los resultados de la farmacoterapia de las leucemias y linfomas. Si bien es cierto que los pacientes pediátricos suelen cumplir adecuadamente los ciclos de medicación oncológica (los padres son plenamente conscientes de la gravedad de la enfermedad y de la importancia del cumplimiento del tratamiento), nunca está de más reforzar la promoción de la adherencia desde la farmacia, especialmente las fases de tratamiento de mantenimiento por vía oral14. Las estrategias para asegurar una implicación activa en el tratamiento deben desarrollarse de forma personalizada, con el paciente y la familia, fomentando su confianza en los fármacos administrados; pueden incluir información verbal y escrita y recursos interactivos, debiendo siempre recordarse que las consecuencias de la falta de adherencia pueden ir desde un empeoramiento de la calidad de vida, una falta de control de la enfermedad y una mayor probabilidad de recaídas, complicaciones o ingresos hospitalarios, hasta la aparición de efectos secundarios o incluso de mortalidad.
Por otra parte, tras una dispensación de inicio o de continuación, y especialmente en los tratamientos prolongados, un adecuado seguimiento farmacoterapéutico (ofrecido por el farmacéutico de forma rutinaria, sistematizada y registrada/documentada, con reuniones periódicas con el paciente) permitirá detectar, atenuar y resolver la posible aparición de resultados negativos y problemas relacionados con la farmacoterapia. La farmacovigilancia ante posibles reacciones adversas (con su correspondiente notificación, en su caso, al Sistema Nacional de Farmacovigilancia), y la identificación y prevención de interacciones farmacológicas y contraindicaciones del tratamiento antineoplásico revertirán en una mejor calidad de vida de los pacientes pediátricos. Además, mediante una actitud vigilante, la detección precoz desde la farmacia comunitaria u hospitalaria de los signos y síntomas que acompañan a una posible recaída o refractariedad a una línea de tratamiento y a las potenciales complicaciones derivadas de la quimioterapia (infecciones, tumores secundarios, etc.) también puede contribuir a activar la ruta asistencial que asegure un cambio de tratamiento temprano. Para todo ello, junto a la recomendación de consultar las fichas técnicas autorizadas de los medicamentos, si se tiene en consideración que la información científica se actualiza constantemente y que recientemente se han aprobado diversas alternativas terapéuticas, cobran especial relevancia las bases de datos que contienen información actualizada y pormenorizada sobre aspectos farmacológicos. Es el caso, por ejemplo, de la base de datos BOT PLUS, que permite, entre otras funcionalidades, la detección y evaluación de interacciones farmacológicas entre múltiples medicamentos y/o principios activos.
Con la incorporación de las primeras terapias CAR-T a la práctica clínica habitual, se abre otro novedoso campo de actuación asistencial para con los pacientes pediátricos oncohematológicos, donde los farmacéuticos hospitalarios pueden jugar un papel crucial (Revuelta et al., 2019), ya que los protocolos farmacoclínicos de fabricación y uso de estos medicamentos, altamente especializados, tienen lugar en el ámbito hospitalario, con intervención también de servicios como
hematología, neurología y medicina intensiva. Pero también será imprescindible la participación de profesionales de atención primaria –entre ellos, el farmacéutico comunitario– en el seguimiento de resultados en salud y la monitorización de la seguridad de estos medicamentos (Figura 11).
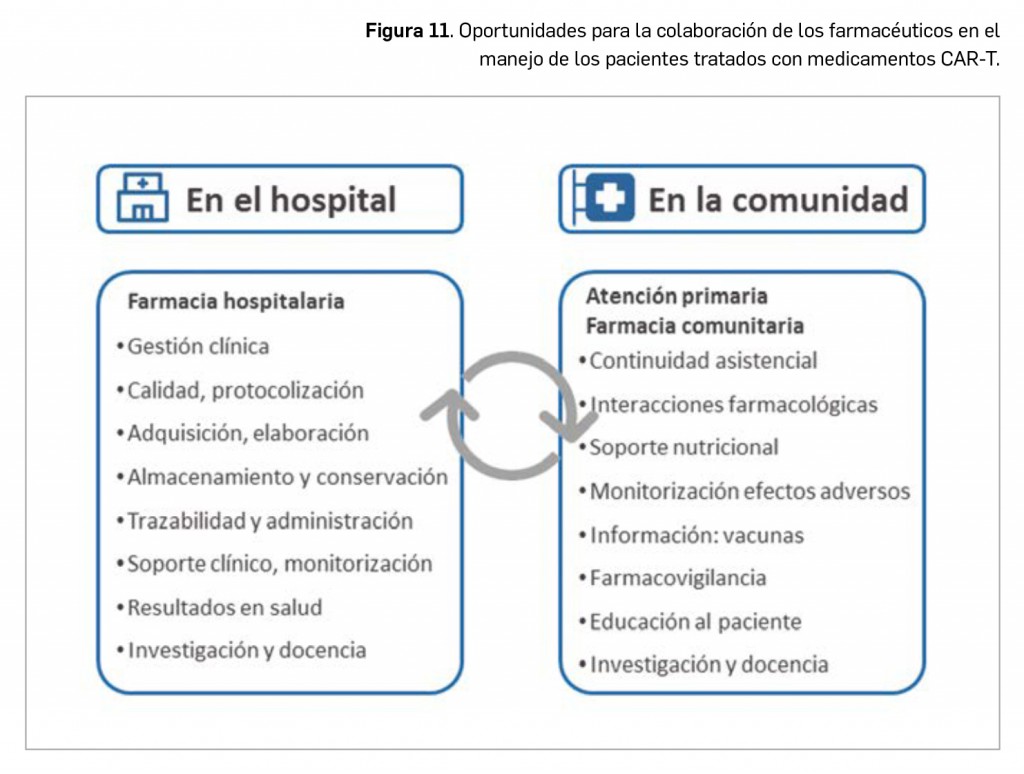
Cuando en un determinado hospital se cursa ante el Ministerio de Sanidad la solicitud de tratamiento de un paciente concreto con estas terapias, y ésta se autoriza, la adquisición de los medicamentos CAR-T comerciales la realizan los Servicios de Farmacia Hospitalaria (SFH) a través de las plataformas que han puesto en marcha los laboratorios y que permiten coordinar los distintos pasos del proceso; los farmacéuticos participan así en a la hora de gestionar la orden de compra en los sistemas de gestión farmacoeconómica. Durante todo el circuito, el equipo que atiende al paciente (hematólogos, farmacéuticos especialistas y otros profesionales sanitarios) debe hacerse responsable de mantener la trazabilidad del producto de la aféresis en los sistemas de información, a través de mecanismos como el código SEC (Single European Code), que permite asegurar la trazabilidad de tejidos y células en toda la Unión Europea, junto a otros identificadores que cada centro considere (número de historia clínica, fecha de nacimiento, etc.). De igual modo, los SFH se implican a la hora de la recepción del medicamento, de forma que se comprueba la identidad del medicamento (fármaco, dosis, SEC de la aféresis, lotes, caducidad, etc.) con los datos que constan en la solicitud; también se comprueban la integridad del producto y las condiciones de conservación durante el envío.
Los SFH garantizarán la seguridad de la administración centrándose en algunos puntos críticos:
Del mismo modo que los pacientes que están en las unidades de hematología y trasplante, quienes reciban medicamentos CAR requerirán terapia de soporte, la cual debe estar protocolizada tanto a nivel hospitalario como extrahospitalario, puesto que puede prolongarse más allá de la estancia en el centro. Algunos ejemplos son la fluidoterapia intravenosa, la nutrición artificial, las heparinas de bajo peso molecular para la tromboprofilaxis en los pacientes encamados, alopurinol o rasburicasa para la prevención del síndrome de lisis tumoral en los pacientes con alta carga tumoral que reciben quimioterapia, antiinfecciosos para la profilaxis de las infecciones bacterianas o fúngicas, y la utilización de los factores estimulantes de colonias para la neutropenia o el manejo de la anemia. A nivel de farmacia comunitaria, el farmacéutico también deberá tener en cuenta que el uso de vacunas con virus vivos atenuados y, sobre todo, de corticoides está contraindicado en estos pacientes, salvo valoración previa por el equipo de hematología. Se ha planteado incluir alertas en los sistemas de prescripción y dispensación asociadas a estos pacientes a lo largo de todo el sistema de salud (atención primaria, especializada, farmacia comunitaria), de forma que se haga una doble verificación previa a la utilización de corticoides.
_______________________________________________________________________________________________
Aunque en otro campo de la investigación biomédica, una breve reseña merece también un artículo publicado por un grupo de científicos israelíes que han pretendido evaluar si un protocolo de terapia con oxígeno hiperbárico (TOH) puede limitar o revertir los procesos biológicos asociados al envejecimiento; se han centrado, específicamente, en el acortamiento de la longitud de los telómeros celulares (regiones de los extremos de los cromosomas que protegen la integridad estructural de los mismos), considerado uno de los eventos centrales del envejecimiento a nivel celular, ligado estrechamente con la senescencia celular, y que determina la pérdida progresiva de capacidad fisiológica en todos los tejidos de nuestro cuerpo.
Trabajos previos habían demostrado que la exposición intermitente y repetida a TOH puede inducir efectos regeneradores que ocurren durante la hipoxia. En el citado estudio, los autores han estudiado el efecto de la exposición a un protocolo concreto de TOH a diario durante 60 sesiones (en un periodo de estudio de 90 días) en 35 adultos sanos e independientes de 64 años de edad o mayores, a los cuales les tomaron muestras de sangre en el inicio, a los 30 y a los 60 días, y tras 1-2 semanas de finalizar el protocolo, con el objetivo de evaluar la longitud de los telómeros y el grado de senescencia en células mononucleares periféricas. Los resultados demostraron que la longitud de los telómeros de los linfotitos T helper, T citotóxicos, natural killer y células B aumentó significativamente en más del 20% de media tras la TOH; el cambio más notable se produjo en las células B, cuyos telómeros crecieron en longitud desde un 25,78% a los 30 días y hasta un 37,6% después de finalizar el periodo de tratamiento (p= 0,007). Se observó, además, un aclaramiento o reducción significativa de un 37,3% en el número de células T helper senescentes (p< 0,0001), y de un 11% en el número de células T citotóxicas tras la TOH.
Así pues, los autores concluyen que la administración de oxígeno puro en el interior de una cámara de presión (hiperbárica) logra detener, e incluso revertir, dos de los principales procesos celulares asociados a la edad. Esto, unido a modificaciones en el estilo de vida o la práctica de ejercicio físico regular, que ya han mostrado algún efecto inhibidor en el acortamiento de los telómeros, puede representar un paso más hacia el reto terapéutico de retrasar el envejecimiento a largo plazo. Se abre la puerta a investigaciones futuras sobre el impacto de la terapia con oxígeno hiperbárico en la reversión de un proceso que, no olvidemos, es fisiológico, pero también a la reflexión sobre las implicaciones bioéticas de este tipo de hallazgos: ¿realmente queremos encontrar la manera de prolongar la vida “indefinidamente”? ¿qué consecuencias sanitarias y socioeconómicas podría conllevar?
A raíz de la publicación de algunos artículos especulativos (por ejemplo, el de Meister et al., 2020) y algunas evidencias clínicas previas con otros tipos de virus respiratorios, desde el inicio de la pandemia se comenzó a plantear en diversos medios la posibilidad de que los enjuagues bucales con ciertos colutorios –con compuestos con actividad antiséptica como cloruro de cetilpiridinio1– podrían ser una medida preventiva eficaz frente al contagio por el SARS-CoV-2; se sugirió que permitiría reducir la carga viral en la cavidad oral a corto plazo y podría ser interesante en situaciones como la asistencia odontológica o médica en pacientes con COVID-19. No obstante, la realidad es que la evidencia disponible es muy limitada y no se pueden extraer conclusiones al respecto.
Recientemente se publicaba un artículo (Meister et al., 2020) que trataba de arrojar luz sobre el asunto. Investigadores de la universidad estatal de Pensilvania (EE.UU.) evaluaron diversos productos comerciales para el enjuague nasal y bucal de composición diversa (por ejemplo, Listerine®, champú de bebés de Johnson & Johnson, Betadine® 5%, etc.), a los que se puede acceder sin prescripción médica; en concreto, estudiaron su capacidad de inactivar los coronavirus humanos en ensayos in vitro con células Huh7 infectadas, un tipo de línea celular derivada de hígado humano. Dichas células fueron expuestas en placas de cultivo a una solución de los colutorios durante periodos de 30 segundos, y 1 y 2 minutos, tras lo cual se midió nuevamente la infectividad del virus. Los principales resultados sugieren que una solución al 1% de champú de bebés para enjuague nasal inactivó más del 99,9% de las partículas virales con un tiempo de contacto de 2 minutos; de igual modo, varios de los colutorios para enjuague de la cavidad bucal, como Listerine® y similares, fueron altamente eficaces en la supresión de la infectividad del virus en más del 99,9% (y hasta del 99,99%) incluso con tiempos de exposición de 30 segundos. Los autores sugieren que esa actividad puede deberse a la inclusión en los diversos productos evaluados de compuestos con actividad antimicrobiana, tales como peróxido de hidrógeno al 1,5%, cloruro de cetilpiridinio, eucaliptol, mentol, metilsalicilato, timol o povidona iodada, entre otros.
Conviene subrayar que dicho artículo presenta grandes limitaciones como para respaldar afirmaciones en el sentido de que los enjuagues bucales/nasales pueden ser eficaces para reducir la transmisión del SARS-CoV-2. La primera y principal se refiere a la propia naturaleza del estudio: un ensayo bioquímico in vitro, que difícilmente puede simular con fiabilidad las condiciones de la mucosa nasal o bucal de los seres humanos. Además, el virus empleado no es el SARS-CoV-2 sino el coronavirus HCoV-229E, causante de resfriado común en humanos, que, si bien pertenece a la misma familia que el primero y puede compartir caracteres genéticos y anatómicos, no permite extrapolar los resultados relativos a su inactivación. Hay que tener en consideración, por último, que el uso excesivamente frecuente o abundante de colutorios puede dañar o irritar la mucosa oral, especialmente aquellos que contienen cierta cantidad de alcohol.
En definitiva, parece evidente que aún se requieren ensayos clínicos prospectivos bien diseñados que demuestren o refuten el papel real que el uso de colutorios y enjuagues nasales y bucales puede tener en la reducción de la carga viral en cavidades que son una potencial fuente emisora o vía de entrada de partículas virales del SARS-CoV-2. Si se confirmara su eficacia, y mientras se espera la inmunización con la vacuna, podría ser una medida profiláctica más (de especial interés en pacientes con COVID-19 activa o en profesionales de alto riesgo), como la distancia social, el lavado de manos o el uso adecuado de mascarillas, dejando claro siempre que esos productos no deben ser ingeridos, ni serán un método radical: un enjuague bucal no protegerá del contagio a través de la vía nasales, y viceversa. Hasta entonces, no debe recomendarse su uso como profilaxis ni, muchos menos, como tratamiento de la COVID-19.
En las últimas semanas se van conociendo algunas revelaciones importantes del Registro clínico a escala nacional SEMI-COVID-19, liderado por la Sociedad Española de Medicina Interna (SEMI), con datos de más de 17.000 pacientes con confirmación de infección por SARS-CoV-2 y que fueron atendidos por casi 900 médicos internistas en 214 hospitales de todo el país. Con los datos de este registro se desarrollan actualmente más de 70 investigaciones que persiguen comprender en profundidad la clínica de la patología.
Uno de los artículos publicados (Rodilla et al., 2020) confirma que la hipertensión arterial es la comorbilidad más frecuente en los pacientes con COVID-19 grave y que se asocia en ellos a un mayor riesgo de mortalidad por cualquier causa, independientemente de otras comorbilidades, sexo y edad. Los autores desarrollaron un estudio retrospectivo, observacional, transversal y multicéntrico, en que analizaron los datos clínicos de 12.226 pacientes (media de edad de 67,5 años, 42,6% mujeres) que fueron ingresados en 150 hospitales españoles, comparando las características clínicas de los que sobrevivieron frente a las de aquellos que fallecieron (N= 2.630, 21,5%) por la enfermedad. Observaron que la hipertensión era la comorbilidad más común (50,9% de los casos) seguida de diabetes (19,1%) y de fibrilación atrial (11,2%). Un análisis multivariable demostró, tras un ajuste por género, edad e índices de riesgo por comorbilidades, que la hipertensión se muestra como un factor significativamente predictivo de todas las causas de mortalidad, con independencia de que fuera tratada previamente con fármacos del tipo IECA o ARA-II (los más ampliamente usados) u otros medicamentos. Destaca el hallazgo, además, de que los pacientes tratados con ARA-II antes de su ingreso presentaban el menor riesgo de mortalidad por todas las causas en comparación con otros antihipertensivos, con una tendencia a mejorar la supervivencia a partir de la segunda semana de estancia hospitalaria. Esto descarta definitivamente las hipótesis planteadas por algunos trabajos al inicio de la pandemia, que sugerían que sería conveniente interrumpir el tratamiento con este tipo de fármacos por el riesgo de potenciar la susceptibilidad y gravedad de la COVID-19.
Otro estudio retrospectivo del SEMI-COVID (Carrasco-Sánchez et al., 2020), en este caso con datos de 11.312 pacientes hospitalizados no críticos, ha demostrado que la hiperglucemia es un fuerte predictor de mortalidad al ingreso hospitalario, con independencia de los antecedentes de diabetes. Del total de pacientes, solo 2.128 (18,9%) tenían diabetes y 2.289 (20,4%) fallecieron durante la hospitalización, situándose la tasa de mortalidad en el 15,7% en el subgrupo de pacientes que presentaban una glucemia de < 140 mg/dl al ingreso, en el 33,7% para aquellos con 140-180 mg/dl, y en el 41,1% para pacientes con > 180 mg/dl de glucosa en sangre. Tras un ajuste por edad, diabetes, hipertensión y otros factores de confusión, los resultados indican que la hiperglucemia es un factor de riesgo independiente de mortalidad (en pacientes con > 180 mg/dl de glucemia basal vs. normoglucémicos HR: 1,50; IC95% 1,31-1,73), que también se asocia con mayor necesidad de ventilación mecánica e ingreso en UCI por COVID-19. Esto sugiere que la determinación de la glucemia y el tratamiento temprano de la hiperglucemia, en su caso, serán recomendables en el manejo de todos los pacientes que sean hospitalizados.
En relación con lo anterior, cabe mencionar brevemente los resultados de otro estudio retrospectivo (Yu et al., 2020) con datos clínicos de 689 pacientes con COVID-19 y diabetes tipo 2 como comorbilidad, procedentes de una cohorte de 3.305pacientes de la ciudad de Wuhan (China). Los autores concluyen que, de forma inesperada, los pacientes diabéticos tratados con insulina tienen una tasa de mortalidad por COVID-19 notablemente mayor (27,2% vs. 3,5%; HR ajustado= 5,38; IC95% 2,75-10,54). Adicionalmente, observaron que el tratamiento con insulina se asociaba a una potenciación de la inflamación sistémica y el agravamiento del daño en diversos órganos vitales, por lo que parece prudente extremar las precauciones cuando los pacientes diabéticos con COVID-19 sean tratados con insulina.
Por último, se puede destacar un tercer estudio (Rubio-Rivas et al., 2020) del SEMI-COVID, derivado de los datos de una cohorte de 12.066 pacientes (58,5% hombres, edad media de 67 años, media de 6,7 días desde el inicio de los síntomas), que plantea que pueden establecerse cuatro grandes grupos fenotípicos de pacientes que ingresan con neumonía por COVID-19, en base a sus características clínicas y sintomatología. Todos ellos comparten la triada de manifestaciones compuesta por fiebre, tos y disnea. El fenotipo más numeroso (8.737 pacientes, 72,4%) era el de pacientes que solo presentaban esa triada; éstos, junto a quienes, además, tenían vómitos, diarrea y dolor abdominal (1.253 pacientes, 10,4%) son los de peor pronóstico y mayor mortalidad, con tasas de mortalidad del 24,1% y el 18,6%, respectivamente. En cambio, los pacientes que presentan síntomas como los de un resfriado común –artromialgias, cefalea, dolor de garganta– (880 pacientes, 7,3%) o con clara pérdida de olfato y gusto (1.196 pacientes, 7,3%) son los de mejor pronóstico (tasas de mortalidad de 4,3% y 14,7%, respectivamente). Estos hallazgos pueden permitir a los clínicos identificar, en base a los síntomas presentes en el momento de la hospitalización, los subgrupos de pacientes con peor pronóstico, e instaurar las medidas de tratamiento más apropiadas en cada caso, profundizando en la idea de una medicina de precisión.
Carrasco-Sánchez FJ, López-Carmona MD, Martínez-Marcos FJ, Pérez-Belmonte LM, Hidalgo-Jiménez A, Buonaiuto V et al. Admission hyperglycaemia as a predictor of mortality in patients hospitalized with COVID-19 regardless of diabetes status: data from the Spanish SEMI-COVID-19 Registry. Ann Med. 2020; 53(1): 103-16. DOI: 10.1080/07853890.2020.1836566.
Queridos lectores,
En estas últimas semanas del año 2020 han sido muchas las noticias aparecidas en diversos medios relativas al avance de la investigación clínica con varios candidatos a vacuna frente a la COVID-19, que nos hacen vislumbrar un futuro a medio plazo más esperanzador que hace unos meses. Es comprensible que la población vea en estas noticias el acercamiento del fin de esta pesadilla. Se comentan en este número de PAM los principales resultados divulgados sobre vacunas, con el objetivo de reflexionar, en última instancia, sobre el propio proceso de comunicación de los resultados –vía notas de prensa– en este contexto extraordinario.
En la situación óptima, se prevé que la Agencia Europea de Medicamentos (EMA) –gracias a los excepcionales procedimientos de evaluación continua de la evidencia que ha implantado– haya autorizado dos vacunas para el primer mes del año entrante, y que puedan empezar a desarrollarse los programas de vacunación poblacional en los distintos países de la Unión Europea durante el primer trimestre; para ello, se deberán afrontar las dificultades logísticas de distribución y mantenimiento de la cadena del frío, en condiciones particulares en algunos casos. Pero, dado que aún faltan varias semanas para iniciar la vacunación de grupos de riesgo, que tardará varios meses en alcanzarse la esperada inmunidad de rebaño que conduzca a la vuelta de la normalidad y, sobre todo, que las cifras de contagios y fallecidos son aún tremendamente altas (muy por encima de lo que una sociedad como la española puede “aceptar”), los profesionales sanitarios debemos hacer un llamamiento a la cautela individual y la prudencia colectiva, en aras de continuar con la tendencia de reducción de incidencia y mortalidad que se ha conseguido en las últimas semanas de noviembre y primeros días de diciembre.
En este periodo hemos conocido, además, datos positivos de los programas de vacunación antigripal que sugieren una cobertura vacunal mayor, y en menores plazos de tiempo, que la registradas en años previos. Si algo positivo puede sacarse de la pandemia que estamos viviendo, quizás la mayor concienciación social sobre temas de salud pública sea una de las mejores lecciones aprendidas. Por otra parte, el Sistema Nacional de Salud ha dado un paso importante hacia el pleno aprovechamiento de sus recursos sanitarios en las estrategias de salud pública: las farmacias comunitarias están dispensando ya test de anticuerpos, calificados de autodiagnóstico, previa presentación de la correspondiente receta médica, y se espera que en algunas Comunidades Autónomas puedan comenzar a realizar test de antígenos –de mayor implicación diagnóstica– en las próximas semanas. No podemos si no mostrar nuestro convencimiento de que avanzar con la mayor rapidez posible en esta línea contribuirá significativamente a mejorar la situación sanitaria.
Entre los contenidos del nuevo número de PAM que les presentamos, el último del presente volumen anual, podemos destacar la revisión monográfica de apertura, que aborda aspectos epidemiológicos y terapéuticos de los principales tumores que afectan a la población pediátrica, los cánceres hematológicos, los cuales presentan, además de su relevancia sanitaria, un importante impacto social y psicológico. Un segundo artículo de revisión complementa a la primera, aportando una visión clínica de otra patología muy frecuente en niños y adolescentes: el trastorno por déficit de atención e hiperactividad. Se incluye también un resumen de la concesión de los Premios Panorama 2020, el galardón con que el Consejo General, a través de esta publicación, premia a los medicamentos más innovadores del año en curso. Cabe destacar que el mes de noviembre transcurrió sin la comercialización en España de ningún nuevo principio activo, lo cual motiva que este número no contenga ninguna evaluación de medicamentos. Sin embargo, en los primeros días de diciembre se comercializaba un nuevo fármaco biológico para el tratamiento de la artritis reumatoide, el upadacitinib. Su evaluación se incorporará en el siguiente número.
Finalmente, mostramos desde aquí nuestro más afectuoso recuerdo para todos los que tristemente han fallecido por la enfermedad infecciosa que marcará en nuestra memoria el difícil 2020 que ahora termina. Con la esperanza de que el próximo año sea notablemente mejor, en todos los sentidos, les deseamos una agradable lectura de este número y que puedan vivir las fechas navideñas que se aproximan con la mayor felicidad posible. Seamos cautos: la vacuna está más cerca, pero el SARS-CoV-2 sigue entre nosotros. Un cariñoso saludo.
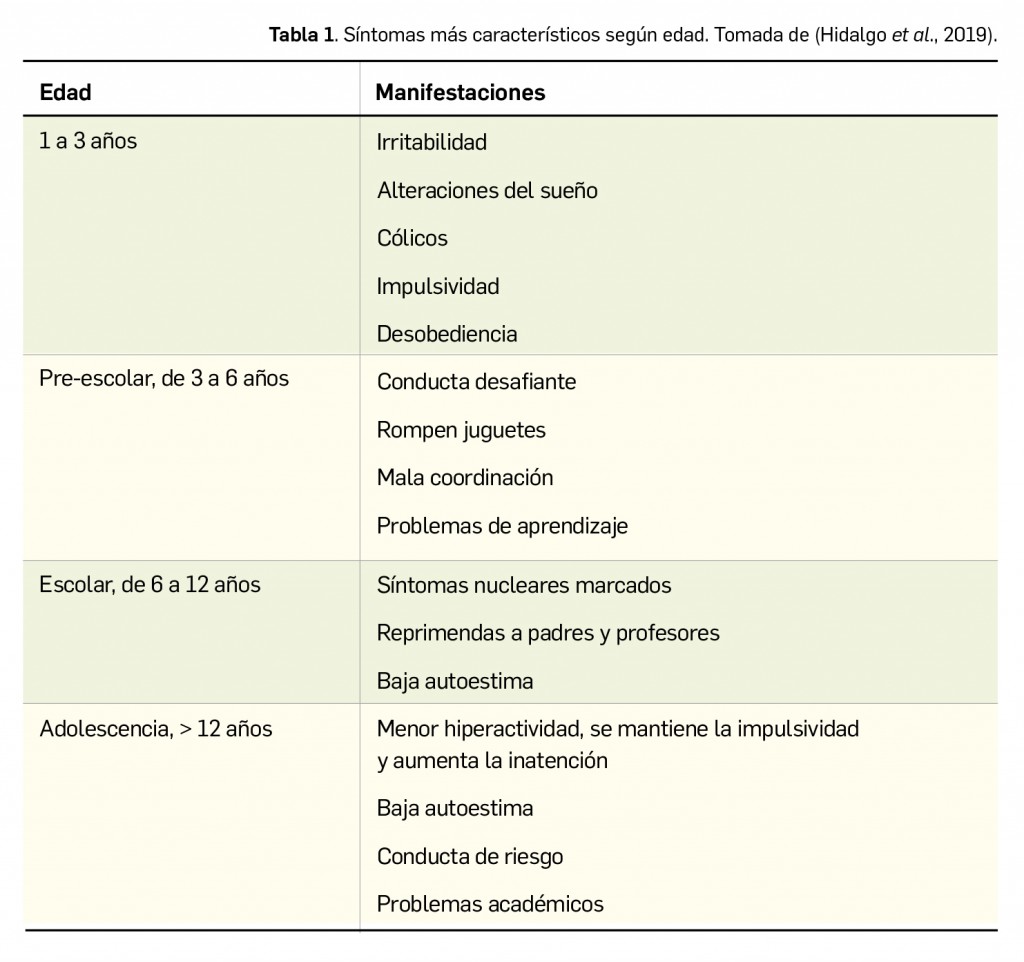
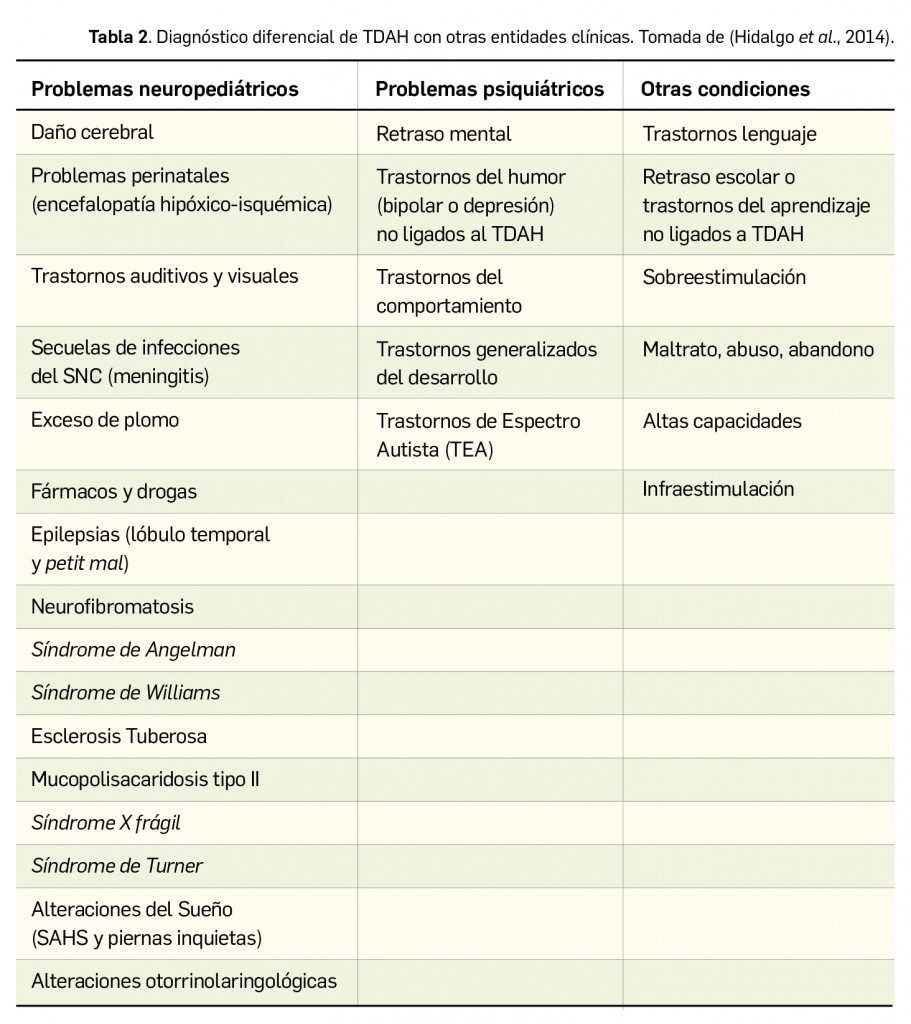
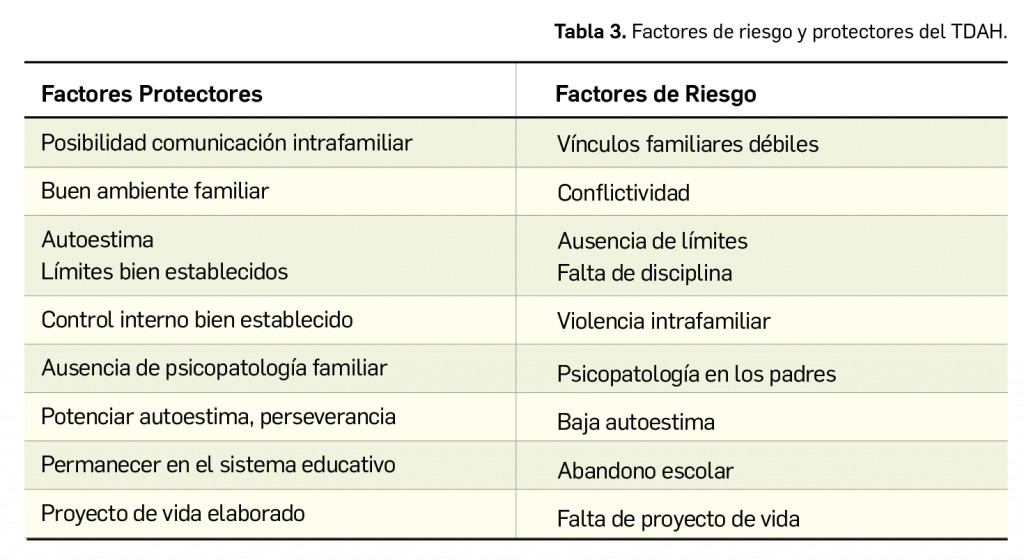
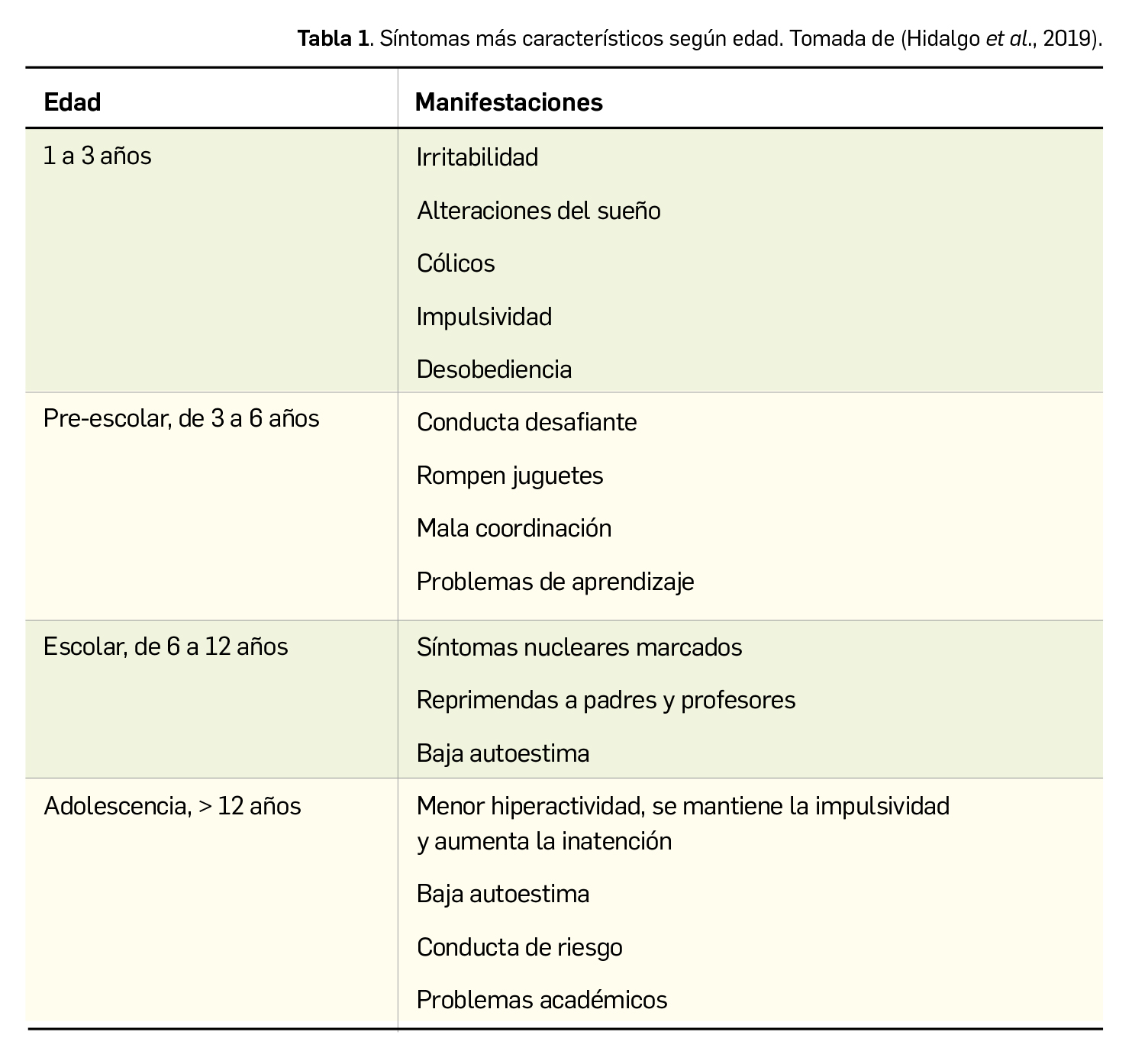{kind=link}
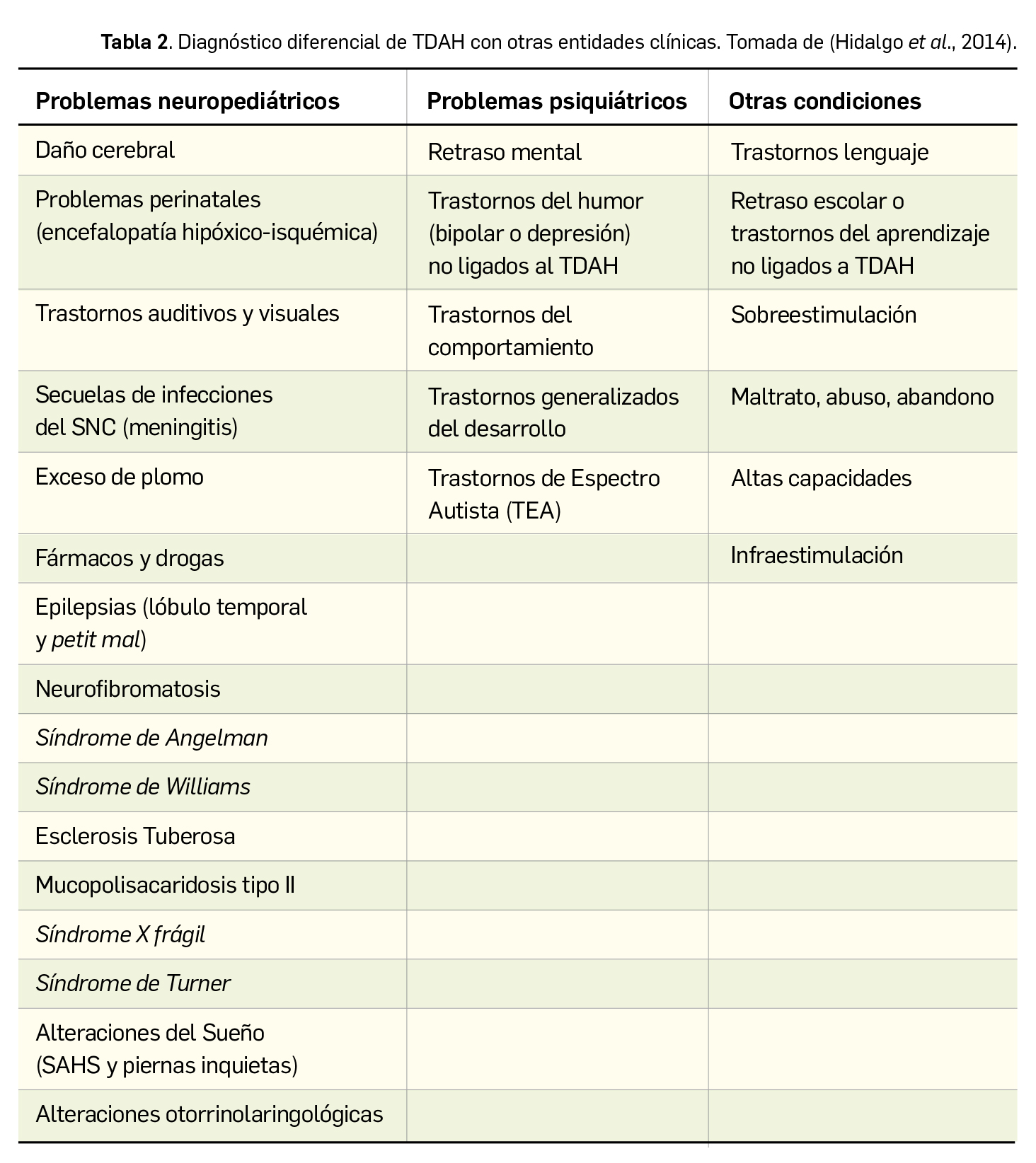{kind=link}
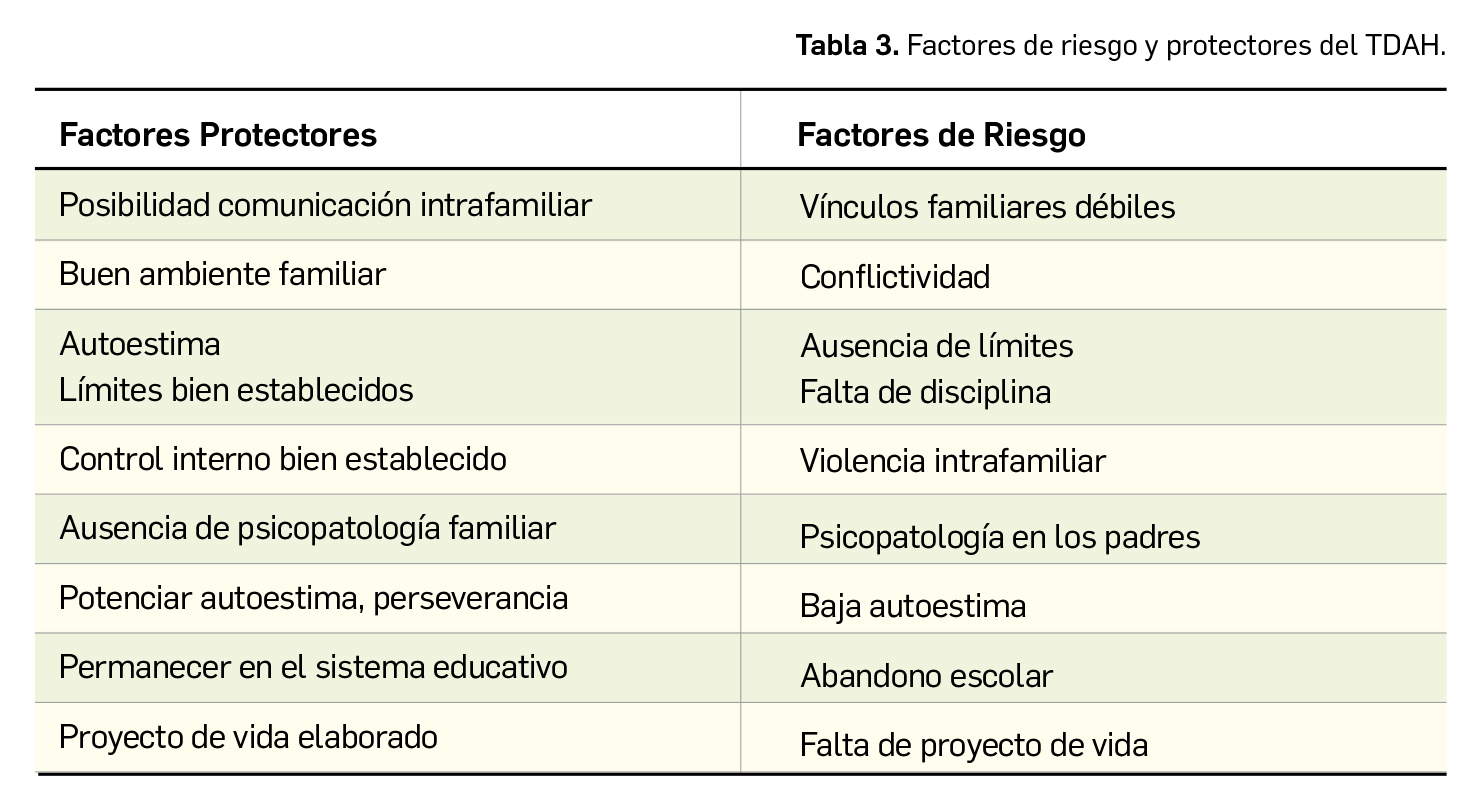{kind=link}