Resumen
El shock es una situación clínica en la que se produce un fallo del sistema circulatorio que conlleva mala perfusión tisular y, por lo tanto, una inadecuada oxigenación y nutrición de las células de los diferentes tejidos del organismo. Supone una amenaza para la vida, que será dependiente directamente al tiempo de actuación, pues los efectos del shock son inicialmente reversibles, pero, si las causas se mantienen, puede progresar a una situación de fallo multiorgánico irreversible y fallecimiento del paciente. Clásicamente se han distinguido 4 tipos en función de su etiopatogenia. En un mismo paciente pueden estar presentes más de uno: hipovolémico, distributivo, cardiogénico y obstructivo. El manejo de una situación de shock, debe ir dirigido al mantenimiento de las constantes vitales del individuo, mediante la administración de drogas vasoactivas (dopamina, dobutamina, norepinefrina) y reposición de fluidos, en tanto se procede lo antes posible a un tratamiento, si fuere posible, etiológico del cuadro.
INTRODUCCIÓN
El shock es un síndrome clínico se caracteriza por la incapacidad del corazón y/o de la circulación periférica de mantener la perfusión adecuada de órganos vitales, bien por bajo flujo sanguíneo, o por una distribución irregular de éste. Independientemente de la causa, el disbalance entre el aporte y las necesidades tisulares de oxígeno y sustratos producen disfunción celular. Si esta situación persiste se acaba produciendo una lesión celular irreversible. Se trata por tanto de un proceso progresivo, con mayor o menor velocidad de implantación. Es un concepto que contrasta con el significado real del vocablo “choque” que tiene un sentido de acontecimiento imprevisto o súbito.
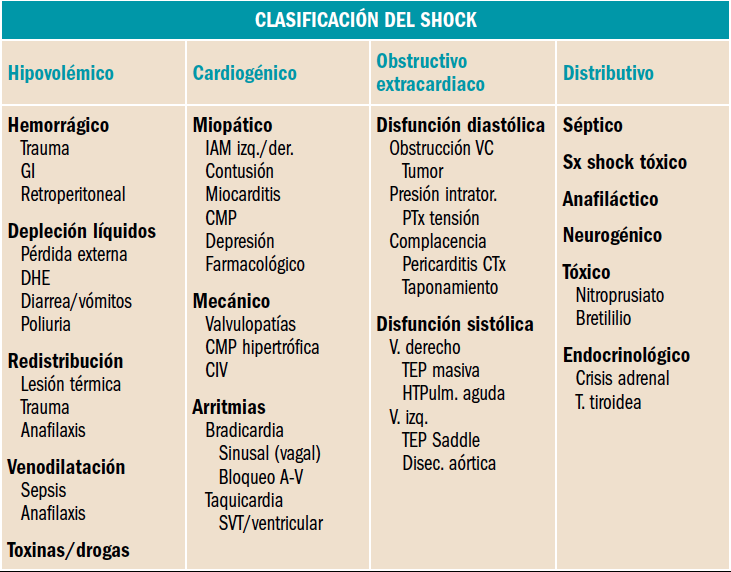
Se ha dividido en cuatro grandes grupos atendiendo a su fisiopatología: cardiogénico, hipovolémico, distributivo y obstructivo o de barrera. Esta clasificación, aunque incompleta, da una visión simplificada, sencilla y didáctica de los mecanismos que concurren en los diferentes tipos de shock.
- AFECTACIÓN DEL CONTINENTE O EL CONTENIDO:
- Shock hipovolémico: Se produce en esta situación una disminución real del volumen circulante (hipovolemia), bien de origen hemorrágico por pérdida súbita y repentina de grandes cantidades de sangre (generalmente pérdidas superiores a 2-2,5 l); o bien por pérdida de volumen plasmático, sin pérdida de sangre. Esto puede ocurrir en pacientes que han sufrido una peritonitis, una sepsis donde se produzca una alteración de la permeabilidad capilar o en grandes quemados. Una tercera situación seria aquella desencadenada por la pérdida de agua y electrolitos, como podría ocurrir ante la presencia de una diaforesis excesiva, vóitos incoercibles, diarreas o ante el uso de dosis altas y continuadas de diuréticos
- Shock distributivo: También denominado vasopléjico, en el que se produce una dilatación vascular excesiva y patológica que impide un adecuado aporte vascular. Es el mismo mecanismo que podría acontecer ante una septicemia, un shock anafiláctico o bien un shock neuorgénico.
- AFECTACIÓN DEL CORAZÓN COMO BOMBA:
- Shock cardiogénico: En este caso lo que se produce es un fracaso del corazón como bomba, en su labor de eyectar la sangre al resto del organismo. Se produce una pérdida de la función contráctil del miocardio. Se puede presentar en episodios de Infarto agudo de miocardio, en situaciones de insuficiencia cardíaca grave de cualquier etiología, tras cirugía cardiaca que implique una lesión miocárdica, o bien ante el mal funcionamiento cardiaco debido a factos mecánicos que desencadenen una situación de sobrecarga sistólica o diastólica grave, como puede ocurrir en lesiones valvulares (insuficiencia aórtica o mitral agudas), rotura del tabique interventricular, o bien ante trastornos severos del ritmo y de la frecuencia (arritmias, taquicardias o bradicardias graves.). Los mecanismos fisiopatológicos finales serían una disminución de la descarga sistólica, una disminución del llenado diastólico y/o alteraciones del ritmo o de la frecuencia cardiaca
- Shock obstructivo: Toda aquella situación que actúe impidiendo una normal eyección cardiaca. Como puede suceder en cuadros de embolismo pulmonar, taponamiento cardíaco, aneurisma disecante de aorta; o bien en situaciones de disfunción de prótesis cardíacas (trombogénicas), obstrucción de venosa importante de venas cavas, neumotórax, y en determinados casos de mixomas auriculares que se comportarían como muy trombogénicos. Fisiopatológicamente se produciría una obstrucción a la eyección del ventrículo, acompañado la mayoría de las veces de una obstrucción al llenado ventricular
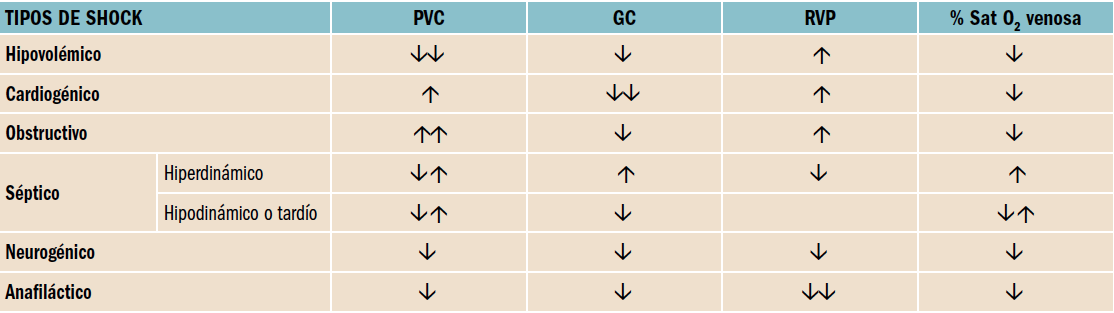
En la tabla se muestran los distintos parámetros hemodinámicos que se pueden alterar en los distintos tipos de shock (PVC: presión venosa central, GC: gasto cardiaco, RVP: resistencia venosa periférica, %Sat O2 venosa: porcentaje de saturación venosa de oxígeno).
SHOCK HIPOVOLéMICO
El mecanismo primario inicial es la pérdida de volumen eficaz circulante. Es un proceso cíclico que una vez desencadenado genera una secuencia de fenómenos cada uno de los cuales afecta desfavorablemente al siguiente. La disminución del flujo sanguíneo a órganos y tejidos vitales causa un suministro insuficiente tanto de O2 como de nutrientes, y también una distribución inadecuada de oxígeno, responsable de las graves alteraciones que genera este estado de insuficiencia microcirculatoria. En el análisis hemodinámico de este tipo de shock aparecen
- Presiones de llenado bajas (baja presión venosa central, presión capilar pulmonar y gasto cardiaco) y
- Resistencias en la microcirculación sistémicas altas.
Dentro de este este tipo de shock podemos hacer a su vez una subdivisión en dos grandes grupos:
Shock hipovolémico hemorrágico
Se presenta como consecuencia de hemorragias internas o externas: laceración de arterias y/o venas en heridas abiertas, de hemorragias secundarias a fracturas; o bien de origen gastrointestinal (úlceras, varices), de hemotórax o de sangrados intraabdominales por rotura de vísceras macizas, complicaciones del embarazo (ectópico), alteraciones de la coagulación.
La disminución de la volemia como consecuencia de una hemorragia aguda puede producir un shock por diminución de la precarga (volumen hemático que recibe el corazón durante la diástole), con lo cual no existe un volumen eficaz que el corazón como bomba pueda manejar y enviar al resto del organismo. La gravedad del cuadro dependerá en gran parte de la cantidad de sangre perdida y de la rapidez con que se produzca. Además hay un gran trasvase de líquido intersticial hacia el espacio intravascular, que es importante corregir para evitar complicaciones.
Shock hipovolémico no hemorrágico
Este cuadro puede producirse como consecuencia de importantes pérdidas de líquido de origen gastrointestinal (vómitos, diarrea), una diuresis excesiva (diuréticos, diurésis osmótica, diabetes insípida), fiebre elevada (hiperventilación y sudoración excesiva), falta de aporte hídrico y extravasación de líquido al espacio intersticial con formación de un “tercer espacio” (quemaduras, peritonitis, ascitis, edema traumático).
SHOCK DISTRIBUTIVO
El shock distributivo es un proceso hiperdinámico donde se produce una vasoplejia sistémica, una vasodilatación excesiva, lo que provoca una hipovolemia relativa, ya que el “continente “ (los vasos) están demasiado dilatados lo que produce que el “contenido” (flujo hemático) sea insuficiente para realizar una perfusión adecuada tisular pudiendo producir daños en el órgano diana.
Aunque la etiología más común es el shock séptico, dentro del que se deben considerar el síndrome de respuesta inflamatoria sistémica (SRIS); Se hace necesario considerar otras etiologías como anafilaxia, reacciones a drogas o toxinas (incluyendo picaduras de insectos), reacción de trasfusión e intoxicaciones por metales pesados, crisis addisonianas, insuficiencia hepática y shock neurogénico debido a una lesión cerebral o de la médula espinal.
Desde un punto de vista hemodinámico nos encontramos con un gasto cardiaco normal o aumentado, unas resistencias vasculares sistémicas disminuidas y una presión de llenado ventricular izquierdo normal o disminuido
SHOCK SéPTICO
Las manifestaciones clínicas del shock séptico son consecuencia de la respuesta inflamatoria del huésped ante la presencia de microorganismos (bacterias, hongos, protozoos y virus) y sus toxinas. Antes de estudiar este tipo de shock es principal conocer y saber diferenciar los siguientes conceptos:
- Infección: Es un término clínico para definir el fenómeno microbiano que se caracteriza por la respuesta inflamatoria a la presencia de microorganismos o a la invasión de tejidos estériles del huésped por dichos organismos.
- Bacteriemia: Se produce por la presencia de bacterias en la sangre. La bacteriemia puede ser transitoria, si dura minutos, intermitente o continua si permanece horas.
- Sepsis: El concepto comprende desde el síndrome de respuesta inflamatoria sistémica (SIRS) a la infección grave documentada, clínica y/o microbiológicamente.
- SIRS: Es una respuesta generalizada del organismo ante determinados estímulos, cuya presencia puede obedecer a causas infecciosas o no infecciosas. Implica la presencia de dos o más de los siguientes: Fiebre > 38 °C o hipotermia < 36 °C. Taquicardia (FC > 90 lpm). Taquipnea >30 rpm, o PaCO2 20 ml/kg en 24 horas. Hiperglicemia en ayunas (glucosa plasmática > 110 mg/dl) en ausencia de diabetes. Niveles plasmáticos altos de procalcitonina o de proteína C reactiva.
- Sepsis grave: Sepsis con disfunción de uno o más órganos (función hemodinámica, renal, respiratoria, hematológica o neurológica) asociada a la sepsis, hipotensión arterial (transitoria o persistente) o hipoperfusión tisular: Hipoxemia con PaFi/FiO2 > 2 mg/dl o incremento > 0,5 mg/dl. Coagulopatía (INR > 1,5 o TTPA 2,0 mg/dl).
- Shock séptico: Hipotensión arterial debida a la sepsis, que persiste y no responde a la expansión del volumen intravascular con líquidos, acompañada de alteraciones de la perfusión (acidosis metabólica o hiperlactacidemia), o requiere de fármacos vasoactivos para mantener la presión arterial.
- Hipotensión debida a la sepsis: Presión arterial sistólica < 90 mmHg, o disminución de la presión arterial sistólica en 40 mmHg o más con respecto a los valores basales, en ausencia de otras causas de hipotensión.
La respuesta sistémica a la infección comienza con la activación del sistema de defensa del huésped, especialmente leucocitos, monocitos y células endoteliales, que juegan un papel central en la amplificación de la cascada inflamatoria. Esta se inicia con la liberación de mediadores solubles, fundamentalmente citoquinas como la interleukina 1 (IL-1) y el factor de necrosis tumoral (TNF-alfa), que activan a su vez el sistema del complemento, la vía intrínseca y extrínseca de la coagulación y la fibrinolísis. Todos ellos en mayor o menor medida participan en la patogenia de la sepsis. El fallo circulatorio del shock séptico tiene un perfil hiperdinámico que se evidencia tras la corrección de la hipovolemia que existe habitualmente y se caracteriza por un GC elevado con disminución de las RVS. Su origen es una vasodilatación marcada a nivel de la macro y la microcirculación. La vasodilatación del lecho arterial tiene un papel central en el fallo circulatorio del shock séptico y es responsable del descenso de las RVS y de la PAM. Otros factores que contribuyen a la hipotensión son la disminución del retorno venoso por venodilatación e hipovolemia. Esta última se presenta de forma secundaria al aumento de la permeabilidad de la barrera endotelial. Esta vasodilatación que no responde a fármacos vasoconstrictores es por sí misma, la causa del fallecimiento de un subgrupo de pacientes con shock séptico.
Existe evidencia de que la producción de NO (óxido nítrico) está muy incrementada en el shock séptico, habiéndose encontrado que la concentración sanguínea de nitritos y nitratos (metabolitos del NO) se encuentra muy elevada. Además, se ha demostrado que existe una relación inversa entre los niveles sanguíneos de estos metabolitos y las resistencias vasculares sistémicas. Estos hallazgos han llevado a la conclusión de que el NO es el principal responsable de la vasodilatación que se produce en el shock séptico. También se ha comprobado que el aumento de la concentración de NO en el músculo liso vascular es la causa de la hiporreactividad (vasoplejia) a las catecolaminas tanto endógenas como exógenas. Por otra parte, en el shock séptico existe una depresión de la función contráctil del miocardio. La determinación de la fracción de eyección ha puesto de manifiesto que la función ventricular está deprimida en todos los casos. Además la ventriculografía isotópica ha demostrado que el VTDVI (volumen total diastólico del ventrículo izquierdo) está aumentado en los pacientes que sobreviven, mientras que es normal en los que fallecen. Se piensa que la dilatación ventricular sería un mecanismo compensador para mantener un volumen de eyección adecuado. Sin embargo, a pesar de que la función ventricular está alterada desde las fases iniciales de la enfermedad, la mayoría de los pacientes con shock séptico mantienen un índice cardiaco normal o elevado, hasta fases avanzadas. La depresión de la función miocárdica se ve compensada por la reducción tan marcada de la postcarga y por la taquicardia habitualmente presente.
La insuficiencia circulatoria que se produce en el shock séptico es consecuencia también del fallo de la microcirculación. En éste concurren al menos tres mecanismos: vasodilatación, microembolización y lesión endotelial. La pérdida del tono vascular impide la autorregulación del flujo sanguíneo a nivel tisular y la adecuada distribución del mismo en los diferentes órganos y tejidos. Además la lesión de las células endoteliales produce un aumento de la permeabilidad capilar y la salida de proteínas al espacio intersticial, por lo que se altera el gradiente oncótico-tisular favoreciendo la formación de edema. Este último aumenta la distancia entre los hematíes y las células y limita la difusión del O2. La lesión de la célula endotelial da lugar a la formación de depósitos de fibrina y microtrombos y favorece el desarrollo de agregados de leucocitos intracapilares. Estas alteraciones de la microcirculación dan lugar a la aparición dentro de un mismo tejido de zonas hiperperfundidas con otras hipoperfundidas en las que se produce hipoxia celular y acidosis láctica.
SHOCK ANAFILáCTICO
Este tipo de shock es consecuencia de una reacción alérgica exagerada ante un antígeno. Son numerosas las sustancias capaces de producirlo. Habitualmente la reacción anafiláctica se produce como consecuencia de la exposición a un antígeno que induce la producción de IgE que se fija sobre la superficie de los basófilos circulantes y sobre los mastocitos tisulares del tracto gastrointestinal y respiratorio y piel que quedan sensibilizados. Cuando la exposición al mismo antígeno se repite, éste se une a las IgE y las activa, iniciándose una serie de eventos bioquímicos que conducen a la liberación de mediadores como histamina, prostaglandinas, factor activador plaquetario, fragmentos de complemento, SRS-A, componentes de la cascada de la coagulación, productos de la vía de la lipooxigenasa y metabolitos del ácido araquidónico. Estos mediadores liberados alteran la permeabilidad capilar a nivel sistémico y pulmonar con formación de edema intersticial y pulmonar. Además hay una vasodilatación generalizada con descenso de la presión arterial y una vasoconstricción coronaria que provoca isquemia miocárdica. También se produce contracción de la musculatura lisa de los bronquios y de la pared intestinal, que causa broncoespasmo, diarrea, náuseas, vómitos y dolor abdominal. La activación de la cascada de la coagulación puede desencadenar una CID. Así pues en la patogénesis de la hipotensión se implican la disminución de la precarga por hipovolémia y vasodilatación, la disminución de la postcarga por descenso de las RVS y la disfunción cardíaca por isquemia.
SHOCK NEUROGéNICO
Este shock es el resultado de una lesión o de una disfunción del sistema nervioso simpático. Se puede producir por bloqueo farmacológico del sistema nervioso simpático o por lesión de la médula espinal a nivel o por encima de T6. Las neuronas del sistema nervioso simpático localizadas en la porción toracolumbar de la médula espinal reciben estímulos cerebrales para mantener los reflejos cardioacelerador y vasoconstrictor. Los estímulos enviados desde el tronco del encéfalo atraviesan la médula cervical y torácica alta antes de abandonar el sistema nervioso central, por lo que un bloqueo farmacológico o una daño medular que interrumpa estos reflejos producirá una pérdida del tono vascular con gran vasodilatación y descenso de la precarga por disminución del retorno venoso, así como bradicardia (que acentúa la hipotensión). El patrón hemodinámico se caracteriza por un GC bajo con descenso de la precarga (PVC: presión venosa central, POAP: presión de oclusión d la arteria pulmonar) y disminución de las RVS.
SHOCK CARDIOGéNICO
El shock cardiogénico es la forma más grave de fallo cardíaco y habitualmente la causa primaria es un fallo de la función miocárdica. Frecuentemente se produce como consecuencia de una cardiopatía isquémica, en la fase aguda de un infarto de miocárdio (IAM), aunque también se ve en la fase final de otras cardiopatías y en diversos procesos patológicos.
El shock ocurre en aproximadamente un 6-8% de los pacientes que acuden al hospital con un infarto agudo de miocárdio (IAM) y la mortalidad derivada de esta complicación supera el 80%. Otros posibles mecanismos de shock en el IAM son el taponamiento cardiaco como consecuencia de la rotura de la pared libre del VI, la perforación septal que da lugar a una comunicación interventricular (CIV), la ruptura aguda de músculo papilar de la válvula mitral y el fallo ventricular derecho.
Existen otras patologías que pueden provocar un shock cardiogénico o contribuir al desarrollo del mismo, como son: disfunción sistólica no isquémica del VI, valvulopatías severas, fallo ventricular derecho, disfunción diastólica del VI, pérdida de la sincronía auriculo-ventricular, taquiarrítmias, bradiarrítmias, fármacos (beta-bloqueantes, calcioantagonistas, quinidina, procainamida…), alteraciones electrolíticas (hipocalcemia, hiperkaliemia, hipomagnesemia), acidemia e hipoxemia severa. Hemodinámicamente el shock cardiogénico cursa con un GC bajo, una presión venosa central (PVC) alta, una presión de oclusión de arteria pulmonar (POAP) alta, incremento de la resistencia vascular sistémica y disminución del trabajo sistólico ventricular izquierdo.
SHOCK OBSTRUCTIVO O DE BARRERA
La principal característica de este shock es la obstaculización del llenado diastólico cardiaco en forma adecuada, debido a la compresión del corazón y de las estructuras circundantes, perdiendo la distensibilidad, produciendo un llenado de la bomba inadecuado. Se puede producir por: Taponamiento producido por sangre o líquido en el interior del saco pericárdico (que es poco distensible), cualquier causa de aumento de presión intratorácica (neumotórax a tensión, protrusión de vísceras abdominales a través de una hernia diafragmática, una presión positiva excesiva en la ventilación mecánica, embolia pulmonar), que obstruya el flujo de salida del ventrículo derecho y altere el llenado ventricular izquierdo. También la existencia de hipertensión arterial pulmonar primaria, tumores intrínsecos o extrínsecos, estenosis mitral o ártica severa, aneurisma disecante de la aorta.
Patrón hemodinámico: Disminución del gasto cardiaco, aumento de la resistencia vascular sistémica y presión de llenado ventricular izquierdo variable.
Cuando se sospecha taponamiento cardiaco, un signo importante es una disminución > 10 mmHg de la PAS durante la inspiración (pulso paradójico).
DIAGNÓSTICO
El diagnóstico de shock es fundamentalmente clínico, basado en la observación de los síntomas y signos que presenta el paciente, así como en la monitorización de éste y la medición de parámetros analíticos directamente relacionados con el proceso. Es importante el reconocimiento precoz del shock, ya que su reversibilidad y, por tanto, su morbimortalidad, dependen del estadio evolutivo en que se encuentre en el momento del diagnóstico. Para establecer el diagnóstico de shock deben estar presentes, al menos, cuatro de los siguientes criterios diagnósticos empíricamente aceptados:
- Apariencia de enfermedad o estado mental alterado
- Frecuencia cardíaca superior a 100 latidos por minuto.
- Frecuencia respiratoria superior a 22 respiraciones por minuto, o PaCO2 inferior a 32 mmHg.
- Déficit de bases en sangre arterial inferior a 5 mEq/l o incremento de lactato superior a 4 mmol/l.
- Diuresis inferior a 0,5 ml/kg/h.
- Hipotensión arterial de más de 20 minutos de duración.
Distinguimos dos tipos de shock en función de que los mecanismos compensadores actúen correctamente o se agoten:
- Shock moderado: Piel: fría, pálida y con retraso en el relleno capilar, intranquilidad, ansiedad, nerviosismo. taquicardia con presión arterial normal o levemente disminuida, taquipnea, oliguria.
- Shock grave: Piel: fría, pálida, cianótica y con livideces en las extremidades, somnolencia, confusión, coma, hipotensión, taquicardia, arritmias, oligoanuria, taquipnea/bradipnea, acidosis metabólica, hipoglucemia.
Es esencial la orientación inicial, de ahí la importancia de que las pruebas complementarias realizadas se dirijan a orientar en el grado de afectación del paciente y en descubrir el origen del shock. Entre dichos estudios, no deberían faltar:
- Hemograma (con recuento y fórmula leucocitaria), importante tanto para conocer la situación inmunitaria del paciente como para orientar en los agentes patógenos responsables del shock séptico (leucocitosis con desviación izquierda en procesos bacterianos, neutropenia en pacientes VIH y en infecciones por Brucella, eosinofilia en parasitosis y shock anafiláctico).
- Hemoglobina con hematocrito, tan importante en los episodios de shock hipovolémico por cuadro hemorrágico así como en los episodios de hemorragia digestiva.
- Estudio de coagulación (plaquetas, fibrinógeno y D-dímero): la plaquetopenia, la disminución del fibrinógeno y la aparición de D-dímero son sugestivos del desarrollo de una coagulación intravascular diseminada (CID), lo que habitualmente refleja una lesión endotelial difusa o trombosis microvascular.
- Bioquímica básica con glucosa, iones, calcio, urea, creatinina, aspartato aminotransferasa (AST, antes llamada TGO), alanina aminotransferas, bilirrubina y lactato.
- Marcadores cardíacos, como troponina I, isoenzima MB de la creatininfosfoquinasa (CPK-MB), mioglobina y marcadores de fallo cardíaco, como los péptidos natriuréticos tipo B (proBNP). Es importante determinar el lactato sérico, ya que, además de su valor diagnóstico, se han correlacionado valores altos (> 2 mmol/l) con aumento en la mortalidad en pacientes con shock séptico.
- Gasometría arterial o venosa: donde se objetivan cambios como la aparición de hipoxemia, acidosis metabólica, consumo de bicarbonato y un exceso de bases negativo.
- Examen de orina: importante en los cuadros sépticos sin foco aparente (cabe tener siempre en cuenta la prostatitis en el varón y la pielonefritis en las mujeres), además de ser el foco de sepsis más frecuente en los pacientes mayores de 65 años.
- Sería importante realizar de forma reglada la proteína C reactiva (PCR) y la procalcitonina (PCT): niveles altos orientan hacia la existencia de una infección sistémica grave y/o bacteriana en lugar de viral o inflamatoria, por lo que son de utilidad para el tratamiento, indicación de antimicrobianos y para valorar la evolución de dichos cuadros. Valores de PCR > 20 mg/l y PCT > 2 ng/ml orientan a infección de origen bacteriano y sepsis grave. En cambio, cifras de PCR < 8 mg/l y PCT < 0,5 ng/ml disminuyen la probabilidad de bacteriemia, con sepsis por debajo del 1-2%.
- Electrocardiograma: habitualmente se observa una taquicardia sinusal, pero puede encontrarse cualquier tipo de alteración del ritmo, así como alteraciones en el segmento ST y onda T debido a las posibles alteraciones iónicas y metabólicas que se dan en todos los pacientes con shock.
- Dado que el shock séptico es el tipo más frecuente que se asiste y trata, es importante intentar realizar un diagnóstico microbiológico mediante hemocultivos.
ESTADIOS
- Shock establecido: Se refiere al paciente que presenta todos los signos de shock, y requiere modificaciones del esquema terapéutico y quizá soporte avanzado de vida.
- Shock compensado: Hace referencia al paciente que recibiendo algún tipo de soporte, mantiene una estabilidad hemodinámica.
- Shock refractario: Paciente que recibiendo el soporte básico y avanzado de vida persiste en estado de shock.
COMPLICACIONES
El shock en fases avanzadas o en condiciones de rápida evolución ocasiona el síndrome de disfunción multiorgánica y afecta a diversos órganos y sistemas:
- Cerebro: encefalopatía metabólica.
- Corazón: síndrome coronario aguda y arritmias.
- Riñón: insuficiencia renal aguda.
- Respiratorio: insuficiencia pulmonar aguda.
- Hematológico: trastornos de coagulación.
- Metabólico: desórdenes electrolíticos y ácido base.
- Nutricional: desnutrición hipercatabólica.
- Gastrointestinal: hemorragia digestiva, hepatopatía aguda.
MEDIDAS DE MANEJO GENERAL
- Oxigenoterapia a alta concentración mediante una máscara con reservorio 15 lts/min y de persistir la hipoxemia cuantificada mediante el análisis de gases arteriales, pulso-oximetría o un trabajo ventilatorio ineficiente se procederá a intubación endotraqueal y uso de ventilación mecánica precoz.
- Fluidoterapia: La resucitación con fluidos se realizara mediante la administración de suero salino inicialmente un volumen de 2.000 a 3.000 cm utilizando dos vías venosas periféricas de 14 G o 16 G, evaluando la respuesta con la monitorización de la presión arterial, frecuencia cardiaca, diuresis horaria, y la presencia de signos de sobrecarga de volumen.
- Fármacos inotrópicos: De continuar la inestabilidad hemodinámica a pesar de una adecuada resucitación con volumen expresada por una adecuada presión venosa central (PVC) o una presión de oclusión de la arteria pulmonar (POAP), debe iniciarse la administración de fármacos vasoactivos:
- Dopamina: Dosis inicial endovenosa de 5 ug/kg/min mediante una bomba de infusión. La administración es continua y se modificará de acuerdo a la respuesta hemodinámica. Dosis máxima de 20 ug/kg/min. Debe ser aplicada por vía venosa central de preferencia
- Norepinefrina: Dosis inicial endovenosa 0,1 ug/kg/min, dosis titulable hasta 15 ug/kg/min. Administrar por bomba de infusión y vía venosa central
- Dobutamina: Dosis inicial endovenosa 2,5 ug/kg/min mediante una bomba de infusión. La administración es continua y se modificará de acuerdo a la respuesta hemodinámica. Dosis máxima 10 ug/kg/min.
MEDIDAS DE MANEJO ESPECÍFICO
Shock Hipovolémico
Se establecen tres categorías o grados de hipovolemia:
Hipovolemia leve (grado I). Corresponde a una pérdida menor de 20% del volumen circulatorio; los fenómenos compensatorios mantienen la PA, pero hay hipotensión postural. La hipoperfusión afecta sólo a ciertos tejidos que la toleran bien, como piel, grasa, músculo esquelético y huesos.
Hipovolemia moderada (grado II). Corresponde a una pérdida de 20-40% del volumen circulatorio. Se afectan órganos que toleran mal la hipoperfusión: hígado, páncreas, bazo, riñones. Aparece la sed como manifestación clínica; puede haber hipotensión en la posición de decúbito dorsal; la hipotensión postural es manifiesta, y hay oliguria y taquicardia leve o moderada.
Hipovolemia severa (grado III). El déficit del volumen circulatorio es > 40%, las manifestaciones de shock son claras y hay hipoperfusión del corazón y del cerebro. Se observan hipotensión, marcada taquicardia, alteraciones mentales, respiración profunda y rápida, oliguria franca y acidosis metabólica.
El tratamiento consiste en la restauración inmediata del volumen circulatorio y del déficit de líquido extracelular, con protección de las vías aéreas mediante:
- Infusión vigorosa de cristaloides en forma de soluciones salinas a través de dos catéteres venosos periféricos de 14 G o 16 G. Tanto el lactato de Ringer (solución de Hartmann) como la solución salina normal son adecuados. Este es el proceso de reanimación, que en general se logra con los primeros dos litros de cristaloides en 30 minutos, si el shock hipovolémico es de origen hemorrágico la localización y control del foco de pérdida son pasos fundamentales para el manejo del shock.
- Transfusiones de sangre total, lo más fresca posible, que provee plasma y componentes hemostáticos.
- Las soluciones salinas hipertónicas han demostrado su utilidad en ciertas condiciones, especialmente cuando es necesario limitar la cantidad total de agua que debe ser infundida.
Shock Cardiogénico
- Fármacos cronotrópicos para corregir bradicardia (atropina, isoproterenol).
- Inotrópicos para optimizar la contractilidad del miocardio (principalmente dopamina y dobutamina).
- Vasodilatadores cuando la resistencia vascular sistémica está elevada.
- Diuréticos para rebajar el volumen circulatorio en presencia de falla congestiva.
- La bomba de balón intra-aórtico es útil en casos debidamente seleccionados.
Shock Distributivo
- Inicio precoz del tratamiento médico (antibioticoterapia) o quirúrgico de la causa del shock distributivo.
- Administración de líquidos intravenosos para mantener el volumen circulatorio. Se prefieren las soluciones cristaloides; usualmente se comienza con 1-2 litros en un período de 30-60 minutos en el adulto; en los niños 10-20 ml/kg. La administración subsiguiente de líquidos depende del estado hemodinámico, a juzgar por la diuresis horaria, la presión arterial y la frecuencia cardiaca;
- Agentes inotrópicos, usualmente dopamina, dobutamina, norepinefrina, si el paciente no responde en cuanto a los valores de la presión venosa central o del gasto cardiaco.
Shock séptico
- Uso racional y adecuado de antibióticos de acuerdo al foco infeccioso inicial, y modificación según resultados de los cultivos.
- Cirugía de manera precoz tras de conseguir la estabilización hemodinámica, especialmente si el foco infeccioso está localizado.
Shock Obstructivo
Aunque la administración de líquidos puede mejorar algo el problema, éste es un fenómeno de carácter mecánico y el tratamiento definitivo es la corrección de la alteración primaria que lo ha causado, drenaje o evacuación pericárdica o permeabilización del vaso obstruido.
bibliografía
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}