Resumen
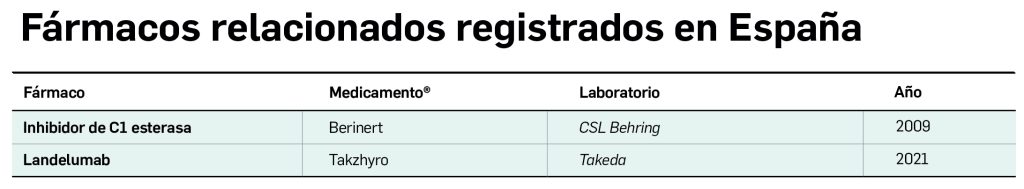
Berotralstat es un nuevo fármaco de molécula pequeña, activo por vía oral, diseñado para actuar como un inhibidor potente y selectivo de la actividad serina proteasa de la calicreína plasmática, una enzima que fisiológicamente participa en la activación del complemento mediante la escisión del cininógeno de alto peso molecular para liberar bradicinina. El potencial terapéutico del nuevo fármaco se explica por su capacidad de suprimir la producción de bradicinina en células endoteliales, de utilidad en pacientes con deficiencia o disfunción de la proteína inhibidora de la C1-esterasa (INH-C1), en quienes está patológicamente aumentada la actividad de la calicreína plasmática y la liberación de bradicinina. Así, el medicamento ha sido autorizado para la prevención rutinaria –terapia oral diaria– de las crisis recurrentes de angioedema hereditario (AEH) en pacientes adultos y adolescentes de al menos 12 años de edad, no pudiéndose usar para el tratamiento de las crisis agudas.
La aprobación de berotralstat en la pauta de 150 mg/día se sustentó en los resultados de un estudio pivotal de fase 3, aleatorizado, multicéntrico, doble ciego, controlado con placebo, de 3 grupos paralelos y diferenciado en 3 partes, que incluyó a 121 pacientes con AEH de tipo I y II. En la parte 1, se demostró que un tratamiento durante 6 meses con el fármaco reducía en un 44 % la tasa mensual de crisis de AEH confirmadas por el investigador (variable primaria) en comparación con placebo, alcanzando significación estadística (p< 0,001). La eficacia, no obstante, fue superior en hombres (reducción del 65 % de la tasa mensual de crisis frente a placebo vs. -29 % en mujeres). En la parte 2 del estudio se confirmó que la eficacia de berotralstat se mantiene a largo plazo (48 semanas): los pacientes tratados con el fármaco todo ese tiempo pasaron de una tasa basal de 3,1 crisis mensuales a 1,1.
En términos de seguridad, el fármaco ha mostrado un perfil toxicológico relativamente benigno. Entre las reacciones adversas más comunes a berotralstat destacan las de tipo digestivo (≈15%; sobre todo dolor abdominal, diarrea y vómitos, en su mayoría pasajeras) así como infecciones del tracto respiratorio superior (incluida nasofaringitis) y cefalea, en su práctica totalidad leves-moderadas. En todo caso, se requieren más datos a largo plazo para esclarecer el perfil beneficio-riesgo en profilaxis prolongada en el contexto de una patología crónica.
Con un mecanismo de acción prácticamente idéntico a lanadelumab, con el que comparte indicación, berotralstat puede ser un tratamiento eficaz y bien tolerado en la prevención de las crisis de angioedema a largo plazo.
Parece que va posicionarse como una opción terapéutica más, alternativa a los tratamientos ya disponibles (sobre todo, en pacientes que no toleren o no estén adecuadamente protegidos con otro fármaco o no hayan sido controlados satisfactoriamente con terapia aguda repetida), respecto a los cuales no parece demostrar ningún beneficio incremental salvo la diferencia de la vía oral (frente a la intravenosa o subcutánea de sus alternativas), que podría impactar potencialmente en una mejor conveniencia para el paciente, aunque no se ha estudiado si mejora la adherencia al tratamiento. La principal guía de práctica clínica europea de la enfermedad recomienda el uso de lanadelumab, los inhibidores de C1 y berotralstat como tratamiento de primera línea para la profilaxis a largo plazo de los pacientes con AEH tipo I o II, sin marcar preferencia de un fármaco sobre otro, de lo que se deduce que berotralstat no va a revolucionar la terapéutica estándar.
Aspectos fisiopatológicos
De forma general, el angioedema se define como una extravasación de líquidos al espacio intersticial que genera una hinchazón de la piel que suele ser autolimitada. Resulta de una pérdida de la integridad vascular que permite que el fluido se desplace hacia los tejidos: la exposición de los vasos a los mediadores inflamatorios provoca la dilatación y el aumento de la permeabilidad de los capilares y vénulas.
El angioedema hereditario (en adelante, AEH) es un trastorno genético de herencia autosómica dominante caracterizado por la aparición episódica de edemas locales subcutáneos y submucosos que afectan fundamentalmente al tracto respiratorio superior y al tracto gastrointestinal. Se trata de una patología rara y grave (potencialmente mortal) que se manifiesta en aproximadamente 1 de cada 60-90 000 individuos europeos1 (algunos autores hablan de una prevalencia en España de 1,09 casos/100 000 habitantes), aunque posiblemente la frecuencia sea mayor, permaneciendo muchos casos sin diagnosticar. Se produce por la falta de regulación de la vía de activación del complemento, siendo clave distinguir el AEH del angioedema inducido por fármacos IECA2.
El angioedema hereditario o familiar existe bajo dos variantes moleculares y fenotípicas, aunque clínicamente indistinguibles. En el de tipo I, que supone el 85 % de los casos, los niveles séricos de la proteína inhibidora de la C1-esterasa (INH-C1, esterasa –serina proteasa– del componente C1 del complemento) están por debajo del 35 % del normal, y se debe al deficiente funcionamiento o alteración del gen SERPING1, localizado en dos cromosomas (el 11 y el 13: 11q11-q13.1), que regula la síntesis de dicha proteína; los pacientes con este tipo de AEH presentan una carencia heterocigota de INH-C1. Por otro lado, en el AEH de tipo II los niveles de INH-C1 son normales o incluso elevados, pero la proteína es estructuralmente anómala y tiene una funcionalidad reducida. Hay otros tipos de AEH minoritarios en que los niveles de INH-C1 son normales: por ejemplo, en el año 2000 se describió un tercer tipo de AEH (tipo III) con niveles y funcionalidad de la INH-C1 normales, para el que se sabe que existen variantes asociadas a determinadas mutaciones (se han identificado mutaciones en genes como el del factor XII de la coagulación, la angiopoyetina-1 o el cininógeno-1) que determinan diferentes síntomas y respuestas a tratamientos; los casos de AEH tipo III en que no se identifica ninguna mutación se conocen como AEH de origen desconocido (AEMPS, 2023).
En condiciones fisiológicas, la proteína INH-C1, perteneciente a la superfamilia de las serpinas, está implicada en la vía generadora de cininas, con una función reguladora tanto para la cascada de coagulación por contacto como para el sistema del complemento. Concretamente, es la responsable de inactivar aproximadamente el 40 % de la calicreína plasmática por unión covalente a esta, de tal forma que una deficiencia funcional o carencia de la INH-C1 provoca el incremento de los niveles de calicreína, que es la principal enzima responsable de la generación de bradicinina a partir de cininógeno. Por otro lado, la INH-C1 actúa como inhibidor del factor XIIa (Factor Hageman activado) de la coagulación, que permitiría la activación de calicreína a partir de su forma precursora en plasma, la pre-calicreína (que está unida al cininógeno de alto peso molecular).
Así pues, la disfuncionalidad o carencia de INH-C1 en ambos sistemas puede contribuir a la formación de edemas, adquiriendo un papel central la bradicinina producida en mayor cantidad: a través de su unión a los receptores específicos B2 de las células endoteliales, es responsable de la intensa extravasación de plasma desde los vasos sanguíneos a la zona tisular lesionada –lo que se traduce en la formación de edema– al tiempo que induce una marcada vasodilatación capilar –sensación de calor y enrojecimiento locales– y contrae la musculatura lisa, produciendo espasmos y dolor. En condiciones normales, la liberación de bradicinina queda inhibida por efecto de la proteína INH-C1, de ahí que su deficiencia o carencia se traduzca en la liberación de una cantidad de bradicinina muy superior a lo normal; este parece ser, en última instancia, el elemento determinante de la formación de edemas y de las crisis del AEH, como manifestación física del aumento transitorio de permeabilidad vascular.
El inicio de las manifestaciones de la enfermedad puede ocurrir a cualquier edad, aunque es más común en la población pediátrica, sin diferencias entre sexos. Los niños afectados de AEH pueden permanecer asintomáticos durante la primera infancia, si bien la mayoría de ellos presenta síntomas desde una edad temprana y las manifestaciones suelen agravarse y aumentar de frecuencia durante la adolescencia. La mayoría de los pacientes con AEH sufren episodios recurrentes e impredecibles de hinchazón transitoria (edema), fundamentalmente de las extremidades, abdomen, cara y vías aéreas superiores. Los síntomas son fluctuantes, manifestándose con ataques o crisis que ocurren espontáneamente a lo largo de la vida del paciente (con frecuencia y gravedad variables3), posiblemente desencadenados por diversas circunstancias o estados, como la ansiedad, el estrés, pequeños traumatismos, cirugía, así como procesos infecciosos. La duración del edema propiamente dicho oscila entre 24 a 72 h.
Por lo general, los episodios de edema son dolorosos o, al menos, molestos e implican limitación funcional. Cuando afectan a la pared del tracto gastrointestinal, resultan en dolor abdominal intenso, náuseas y vómitos y, en algunos casos, diarrea líquida. Los que afectan a la cara y la garganta son particularmente graves y requieren tratamiento inmediato, ya que el edema laríngeo (hasta el 0,9 % de todas las crisis) que cursa con inflamación puede impedir el aporte de aire y causar potencialmente la muerte por asfixia; más de la mitad de los pacientes presenta alguna crisis de este tipo a lo largo de su vida y es típico que la inminencia de un edema laríngeo venga indicada por la dificultad al tragar y un cambio en el tono de voz. Además, el AEH ha sido relacionado con alteraciones linfoproliferativas de células B a largo plazo, tales como la leucemia linfocítica crónica, mieloma múltiple o crioglobulinemia esencial (Gómez et al., 2014).
Los enfermos de AEH son particularmente vulnerables a la cirugía dental, lo que obliga a adoptar medidas precautorias especiales en pacientes con este tipo de historial. En las mujeres, la menstruación y el embarazo parecen incidir significativamente en la patología, aumentando la frecuencia de los ataques; el uso de anticonceptivos orales también está asociado con una mayor frecuencia y gravedad de los ataques, lo que contraindica su uso en estas pacientes. Por otro lado, algunos pacientes experimentan síntomas a modo de aura –cosquilleo o tensión en la zona–, sugerente de un próximo ataque desde 30 min a 24 h después de su aparición. Asimismo, una parte de los pacientes (25 %) con AEH sufren una erupción eritematosa generalizada antes y durante los ataques.
El sistema del complemento
Distribuido entre el plasma sanguíneo y la superficie celular, consiste en un complejo conjunto formado por una veintena de proteínas bioactivas (enzimas proteolíticas, proteínas inflamatorias, receptores de superficie y proteínas con capacidad de provocar lisis celular) que se caracteriza por una estrecha regulación. Es un sistema proteico capaz de lisar células por inserción de sus componentes en las membranas, cuya activación en cascada (y la interacción entre los elementos a que dan lugar) condiciona numerosas funciones efectoras de la inmunidad y la inflamación.
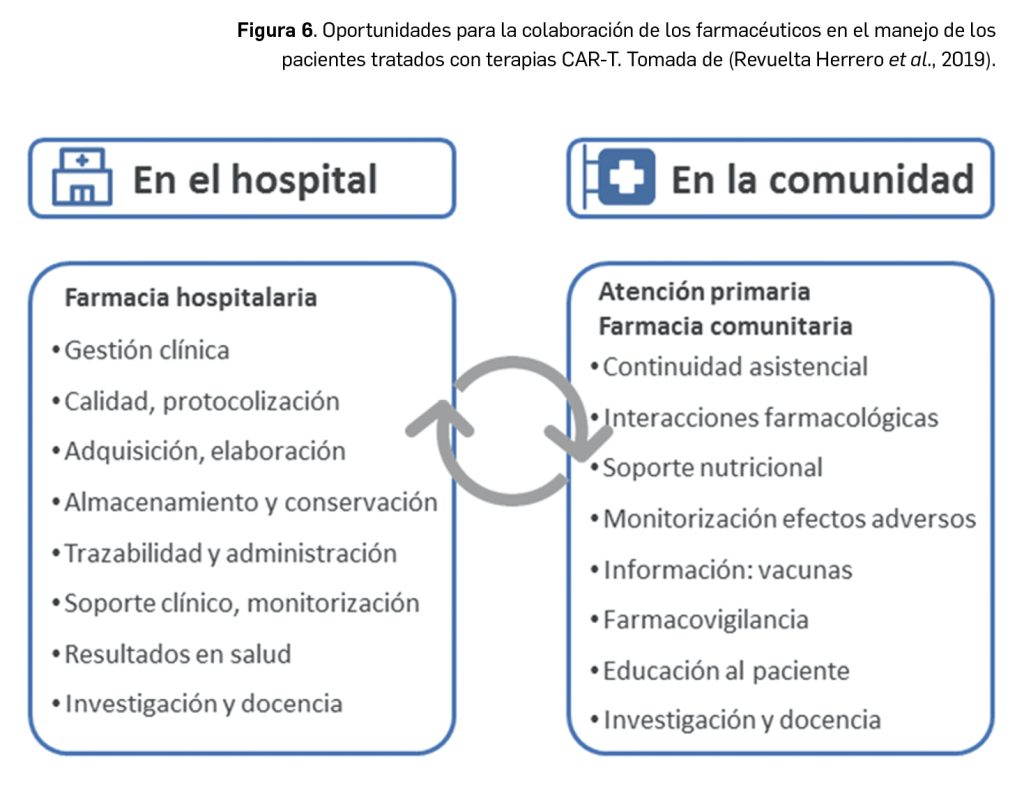
En condiciones fisiológicas, el complemento actúa en los procesos inmunitarios de defensa frente a microorganismos, y su objetivo final es la lisis de los mismos mediante la formación del denominado complejo de ataque de membrana (CAM). El paso más importante para su función biológica es la activación de su tercer componente, C3. La formación de la proteasa responsable de la fragmentación del C3 se puede producir por 3 posibles vías (Figura 1): a) vía clásica, que se inicia por la activación del C1 sobre el anticuerpo (IgG o IgM) unido al antígeno extraño; b) vía alternativa, cuya activación es espontánea o bien consecuencia de la activación previa de la vía clásica, a la que amplifica; y c) vía de las lectinas, incluida frecuentemente dentro de la “vía alternativa”, y que se activa cuando la lectina ligadora de manosa (MBL) reconoce azúcares bacterianos.
Cada una de las vías de activación del complemento cuenta con elementos reguladores. La vía clásica es bloqueada por el inhibidor de la C1-esterasa (INH-C1), la proteína que se une a C4, y el factor I, mientras que la vía alternativa es regulada por el properdina, el factor H o factor acelerador de la degradación de complejo C3C, Bb y el factor I. En todo caso, con independencia de cuál sea la vía de activación de la C3 convertasa, ésta divide al C3 en dos fragmentos: C3a, que es liberado, y C3b, que continúa la cascada de activación y dará lugar a C5 convertasa, la cual, a su vez, interacciona con C5 para liberar C5a y C5b. Este último (C5b) se incorpora a la formación del CAM (C5b-9): cuando el CAM se une a los extremos hidrofóbicos de la bicapa lipídica de las células diana forma finalmente canales transmembrana que provocan la lisis celular.
En resumen, el complemento funciona como una cascada proteolítica en la que un componente activado escinde al siguiente, dando lugar a dos fragmentos activos, uno de mayor peso molecular que se fija a la superficie activadora, y otro de menor peso molecular con función quimiotáctica. Los fragmentos pesados C3b y C5b son necesarios para la formación de poros en la superficie activadora, y los ligeros, como el C3a y C5a (anafilotoxinas), se unen a receptores en mastocitos y basófilos, y provocan la liberación de histamina y otros mediadores anafilácticos. El C5a actúa, además, como quimioatrayente para neutrófilos y monocitos.
En la terapéutica del AEH se pueden identificar tres enfoques o estrategias diferentes, que se resumen a continuación. El objetivo terapéutico, tanto de la profilaxis como del tratamiento agudo, consiste en evitar o disminuir al máximo las crisis de edema, sobre todo si afectan a las vías respiratorias superiores, así como mejorar la calidad de vida de los pacientes (AEMPS, 2023).
-Para el tratamiento sintomático de las crisis agudas ya instauradas es prioritaria la protección de las vías respiratorias, pudiendo ser necesaria la intubación y, en casos muy graves, la traqueotomía. Si bien en los años 2000 se podía emplear plasma fresco congelado, a día de hoy las guías clínicas contemplan de forma indiferente el tratamiento de elección en los cuadros graves mediante la administración intravenosa de concentrado plasmático –derivado de plasma humano– de inhibidor de la C1-esterasa (Cinryze® o Berinert®) o la administración subcutánea del otro fármaco autorizado en esta indicación, icatibant4 (Firazyr®), que normalmente logran reducir el angioedema en 30 min-2 h (y una completa remisión de los síntomas en 24 h); presentan tasas de respuesta que oscilan entre el 65 % y 80 %. Además, la forma intestinal del ataque requiere un tratamiento analgésico intenso, así como una terapia de rehidratación apropiada.
-Más recientemente se ha incluido conestat alfa entre las opciones disponibles para el tratamiento agudo de las crisis de AEH. Se trata de un análogo recombinante de INH-C1, producido en leche de conejas transgénicas, de uso por vía intravenosa en adultos, adolescentes y niños (a partir de 2 años) con AEH debido a un déficit de inhibidor de la C1-esterasa. Con tasas de respuesta parecidas a las del derivado plasmático, demostró eficacia significativa en la reducción del tiempo hasta el inicio del alivio de los síntomas de aproximadamente 120 min frente a placebo, y en el tiempo hasta la aparición de síntomas mínimos de unos 850 min.
-Los pacientes con diagnóstico de AEH establecido –con confirmación bioquímica– pueden requerir una profilaxis a corto plazo en determinadas circunstancias, especialmente cuando vayan a ser sometidos a procedimientos quirúrgicos o dentales, o ante otras situaciones y/o eventos anticipados que puedan precipitar un ataque. En este supuesto concreto, en que antes se solían emplear fármacos androgénicos atenuados (por ejemplo, danazol5) y antifibrinolíticos (por ejemplo, ácido tranexámico), ahora se prefiere el inhibidor de la C1-esterasa.
-Por último, tras una exhaustiva valoración individual (considerando calidad de vida y preferencias del paciente), puede ser necesaria una profilaxis a largo plazo en aquellos pacientes que experimentan un ataque o más de angioedema, a fin de disminuir el número total y la gravedad de las crisis. Hasta hace poco, en España solo tenían esta indicación aprobada el inhibidor de la C1-esterasa –Cinryze® intravenoso y Berinert® subcutáneo6–, en pauta de dos dosis semanales (una cada 3 o 4 días), y todavía en ocasiones se recomienda el tratamiento de 1ª línea con antifibrinolíticos en menores de 18 años y andrógenos atenuados en adultos y valorar, en caso de no mejoría o efectos secundarios, la administración de concentrado plasmático INH-C1.
Hace poco se incorporó lanadelumab, un anticuerpo monoclonal humano que se une específicamente a la calicreína plasmática e inhibe de forma dosis-dependiente su actividad proteolítica, tanto en su forma soluble como unida a membrana, de modo que reduce la proteólisis de cininógeno y la subsiguiente liberación de bradicinina: el control sostenido de lanadelumab sobre la generación de bradicinina limita el efecto de ésta sobre sus receptores B2, reduciendo la inflamación y la extravasación. El nuevo medicamento, designado como huérfano, ha sido autorizado para su administración por vía subcutánea cada 2-4 semanas en la prevención rutinaria de las crisis recurrentes de AEH en pacientes a partir de los 12 años. En los ensayos clínicos, un tratamiento de 26 semanas demostró ser significativamente más eficaz que placebo al reducir la tasa de ataques mensuales en pacientes con AEH en hasta un 87 %; también redujo el número de crisis que precisaron tratamiento agudo, aumentando notablemente la proporción de pacientes sin crisis durante el tratamiento (31-44 % vs. 2 % con placebo).
En cualquier caso, pese al tratamiento preventivo a largo plazo con estos fármacos, algunos pacientes continúan padeciendo ataques severos frecuentes, por lo que es interesante la incorporación de nuevas opciones farmacológicas.
Acción y mecanismo
Berotralstat es un inhibidor sintético de la calicreína plasmática activo por vía oral: es capaz de reducir la actividad serina proteasa de la calicreína, enzima de la vía de activación del complemento que escindiría el cininógeno de alto peso molecular para liberar bradicinina (potente vasodilatador que aumenta la permeabilidad vascular). Se comprende, por tanto, que puede tener potencial terapéutico en pacientes con deficiencia o disfunción del INH-C1, en quienes está patológicamente aumentada la actividad normal de la calicreína plasmática y la producción de bradicinina, causante último de las crisis de edema. Así, el medicamento ha sido autorizado para la prevención rutinaria –terapia oral diaria– de las crisis recurrentes de angioedema hereditario (AEH) en pacientes adultos y adolescentes de 12 años en adelante, no pudiéndose usar para el tratamiento de las crisis agudas de AEH.
Los estudios pre-clínicos han probado que berotralstat ejerce una inhibición potente sobre la calicreína plasmática (Ki= 44 nM) y de aceptable selectividad respecto a otras serina proteasas relacionadas (CI50= 0,88 nM para calicreína vs. 3967 nM para plasmina y > 50 000 nM para otras proteasas); si bien no se puede descartar su efecto in vivo sobre otras dianas similares, no parece que esto tenga consecuencias clínicas. También se ha demostrado que el fármaco suprime la producción de bradicinina en células endoteliales humanas con una CE50 de 5,6 nM tras la activación del sistema de la coagulación, lo que respalda su mecanismo de acción en AEH (EMA, 2021).
Aspectos moleculares
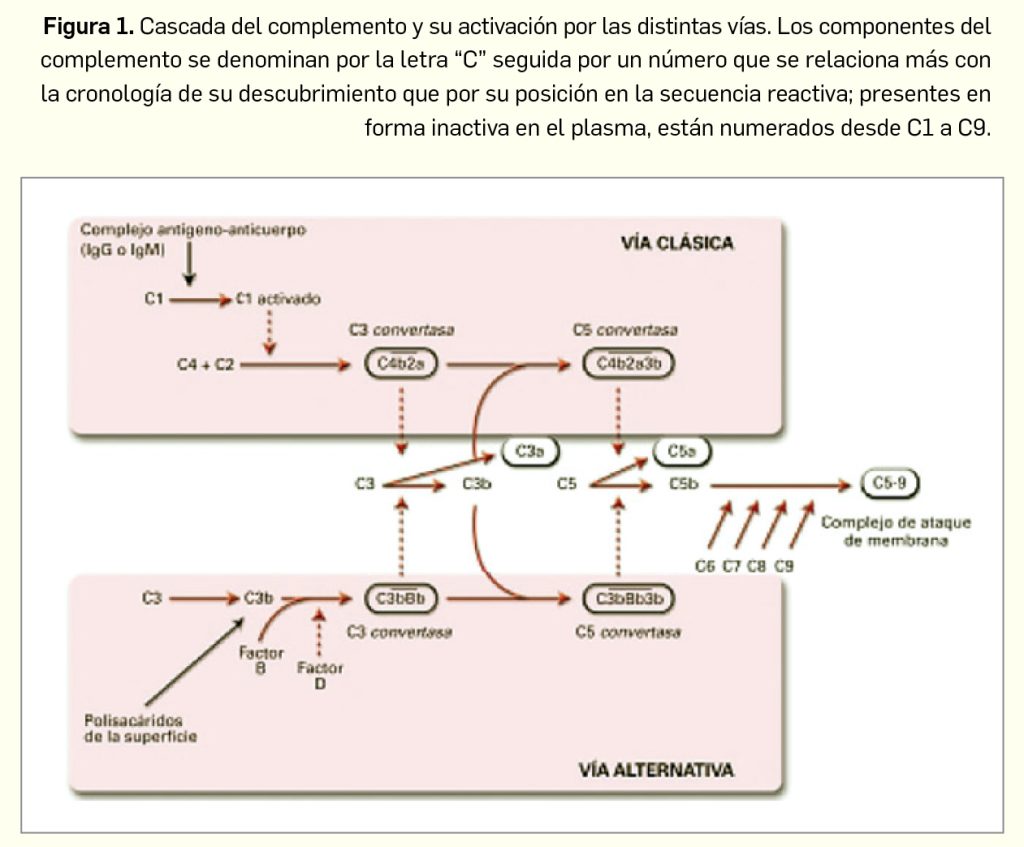
El nuevo fármaco es una molécula sintética relativamente pequeña (Figura 2) obtenida racionalmente mediante una estrategia de “diseño guiado por estructura” (Kotian et al., 2021). Tiene por nombre químico el (R)-1-(3-(aminometil)fenil)-N-(5-((3-cianofenil)((ciclopropilmetil)amino)metil)-2-fluorofenil)-3-(trifluorometil)-1H-pirazol-5-carboxamida, y se presenta en forma de su sal dihidrocloruro, correspondiéndose con la fórmula C30H26F4N6O·2HCl y con un peso molecular relativo de 635,5 g/mol.
El principio activo es un polvo higroscópico blanquecino con solubilidad dependiente del pH. La molécula exhibe estereoisomería por la presencia de un centro quiral, siendo el isómero R la forma activa.
Eficacia y seguridad clínicas
Los datos de eficacia y seguridad de berotralstat en la pauta aprobada (150 mg en una toma oral diaria) para la profilaxis de crisis de angioedema hereditario derivan fundamentalmente de un ensayo clínico pivotal aleatorizado de fase 3 (estudio APeX-2), multinacional y multicéntrico, de grupos paralelos, doblemente ciego y controlado por placebo, en que participaron 121 pacientes con diagnóstico confirmado de AEH7 de tipo I y II (114 adultos y 6 niños de > 12 años).
Para su inclusión se debía confirmar que los pacientes habían sufrido, en las 8 primeras semanas antes de su aleatorización y la primera dosis (periodo de pre-inclusión en que se estableció la tasa basal de crisis), al menos 2 crisis de AEH separadas ≥ 48 h y requeridoras de tratamiento o causantes de deterioro funcional. A partir de ahí se asignaron al azar (1:1:1) a recibir berotralstat a una de las dos dosis evaluadas de 110 mg/día (n= 41) y 150 mg/día (n= 40) o un placebo equivalente (n= 40) y el estudio se dividió en 3 partes con objetivos diferentes: i) parte 1, que buscó evaluar la eficacia de las dos dosis frente a placebo durante 24 semanas; ii) parte 28, investigó también seguridad y tolerabilidad a largo plazo del fármaco, sin placebo como control, desde la semana 24 a la 48; y iii) parte 3, con el mismo objetivo que la anterior, pero en un periodo abierto de extensión entre las semanas 48 y 96 de tratamiento.
Las características demográficas y clínicas de los participantes se balancearon adecuadamente entre grupos de tratamiento, salvo por sexos. Sobresale que un 95 % de los pacientes eran adultos (7 % mayores de 65 años) con una media de edad de 42 años, el 93 % de raza blanca (el 72 % procedía de Norteamérica) y una mayoría eran mujeres (58 % en el brazo de berotralstat 150 mg). Además, en torno a la mitad de los pacientes (52 %) había debutado con manifestaciones de la enfermedad antes de los 12 años (33 % entre los 12 y los 17), tuvieron una media de edad al diagnóstico de 20 años, el 84 % tenía antecedentes familiares de AEH y tres de cada cuatro pacientes había usado medicación profiláctica previamente; las regiones anatómicas donde se notificaban con mayor frecuencia las crisis al inicio eran estómago/abdomen (97 %), manos/brazo (93 %) y pies/piernas (88 %), si bien una alta proporción también reportó haber tenido afectación laríngea (74 %; media de casi 10 crisis laríngeas a lo largo de la vida).
La variable principal de eficacia fue la tasa de crisis de AEH confirmadas por el investigador en un periodo de 28 días tras 24 semanas de tratamiento, mientras que como objetivos secundarios más relevantes se consideraron los efectos de berotralstat en la calidad de vida (medidos por el validado Cuestionario de calidad de vida en pacientes con angioedema AE-QoL y el cuestionario de satisfacción con la medicación TSQM) así como en el número y proporción de días con síntomas de angioedema.
Por ahora se han divulgado los resultados derivados del análisis por intención de tratar de la parte 1 (Zuraw et al., 2021), que fue el conjunto de datos evaluados por la EMA y de la parte 2 (Wedner et al., 2021). Los análisis de sensibilidad han respaldado los resultados del análisis primario.
Si se considera que la mediana de la frecuencia basal de crisis era de 2,9 al mes, resulta estadística y clínicamente significativa la reducción que frente a placebo determinó el tratamiento con berotralstat 150 mg/día en la parte 1: 1,31 crisis mensuales de AEH tras 24 semanas de tratamiento frente a 2,35 crisis con placebo, suponiendo una reducción porcentual del 44 % frente al control (IC95 % 23,0-59,5; p< 0,001). Cabe destacar que la diferencia sustancial entre tratamientos se observa desde el primer mes y se mantiene hasta el final del periodo evaluado en esta primera parte del estudio (6 meses). Más de la mitad de los pacientes que recibieron el fármaco (58 %) redujo la frecuencia de crisis en ≥ 50 % respecto al valor basal, frente a solo un cuarto (25 %) en el grupo de placebo; con respecto a la frecuencia de crisis que requirieron tratamiento agudo9, la reducción con la dosis alta del fármaco fue del 49 % en comparación con placebo (frecuencia en 28 días de 1,04 frente a 2,05). El análisis por subgrupos no reveló diferencias notables de eficacia del fármaco –la reducción de la tasa de crisis era consistente– según factores como la edad (incluidos los pacientes adolescentes) o la frecuencia basal de ataques, pero se observaron diferencias por sexos: los hombres alcanzaron una mayor reducción en la tasa de crisis de AEH (-65 % con berotralstat 150 mg/día frente a placebo) que las mujeres (-29 % frente a placebo).
El beneficio con la dosis de berotralstat de 110 mg/día fue menor: la tasa de crisis de AEH confirmadas en un mes se redujo en un 30 % respecto a placebo al final de los 6 meses de tratamiento, sin alcanzar significación estadística.
Entre las variables secundarias, destaca el efecto positivo observado con el nuevo fármaco sobre la calidad de vida según puntuación del cuestionario AE-QoL, reflejado en una reducción a la semana 24 de -14,6 puntos10 con la pauta autorizada que, no obstante, no alcanzó significación estadística en la comparación con placebo (-9,7 puntos; diferencia media entre brazos de -4,9 puntos, IC95 % -12,2 a 2,4; p= 0,19); la mejora más pronunciada se apreció en la puntuación del dominio de funcionamiento (diferencia de -9,1 puntos entre brazos), en mayor medida que en los de miedo/vergüenza (diferencia de -5 puntos), nutrición (-2,7 puntos) y fatiga/estado de ánimo (-2,2 puntos). Asimismo, la puntuación del grado de satisfacción global reportado por los pacientes según el cuestionario TSQM también se mostró favorable para el nuevo tratamiento: tras 24 semanas se vio una diferencia media respecto a placebo de 18,9 puntos (p= 0,0111).
Otras variables secundarias revelan, por ejemplo, que la pauta autorizada de berotralstat, siempre comparada frente a placebo, se tradujo en una reducción de aproximadamente 13 días más sin síntomas de angioedema en los primeros 6 meses (p= 0,006) o en una reducción del 47 % en la tasa de crisis de AEH confirmadas por el investigador durante el periodo de tratamiento efectivo, esto es, tras alcanzar el fármaco el estado estacionario (p< 0,001). Se redujo también la tasa de crisis de AEH a nivel abdominal, periférico y laríngeo, así como la incidencia de crisis moderadas y graves, y se verificó una disminución del uso de medicación para el tratamiento de las crisis agudas (AEMPS, 2021; AEMPS, 2023).
En la parte 2 del estudio, los pacientes que completaron las 48 semanas de tratamiento con la pauta aprobada del fármaco pasaron de tener 3,06 crisis mensuales al inicio a tener 1,06 crisis tras casi un año de tratamiento, o sea, una reducción del 67 %; la reducción fue algo menor –del 52 %– con la dosis de 110 mg/día (de 2,97 a 1,35). Así pues, parece evidente que la mejora de la frecuencia de crisis observada en la primera parte del estudio continuó en la segunda, demostrando una clara durabilidad de la respuesta al tratamiento con el nuevo fármaco. Entre los pacientes realeatorizados de placebo a berotralstat 150 mg/día al inicio de esta parte, la tasa media de crisis mensuales se redujo de forma constante y consistente (hasta 0,57 crisis/mes a la semana 48).
En cuanto a las variables secundarias, se vio que los pacientes que continuaron en terapia con el fármaco mantenían la mejoría en la puntuación total del cuestionario AE-QoL desde la semana 4 y hasta la 48. E, igualmente, los pacientes realeatorizados de placebo a berotralstat vieron reducir –mejorar– sus puntuaciones totales de AE-QoL al iniciar el tratamiento, con dos tercios (67 %) alcanzando la mínima diferencia clínicamente importante en la semana 48, y mantuvieron la mejoría en el grado de satisfacción con el tratamiento según puntuación del cuestionario TSQM.
Adicionalmente, se han publicado los resultados de soporte de otro ensayo clínico de fase 3 (estudio APeX-J), realizado en Japón con un diseño similar al pivotal (aleatorizado, doble ciego, controlado por placebo y de grupos paralelos), en 3 partes diferenciadas, y también criterios de inclusión y variables semejantes. Enroló a 19 sujetos de ≥ 12 años con AEH tipo I y tipo II (95 % de raza asiática, 84 % mujeres y edad media de 42 años), quienes fueron asignados al azar (1:1:1) a recibir berotralstat 110 mg/día (n= 6) y 150 mg/día (n= 7) o un placebo equivalente (n= 6).
El análisis por intención de tratar de los datos obtenidos en la parte 1 del estudio (Ohsawa et al., 2021) ponen de manifiesto que el tratamiento durante 24 semanas con el fármaco en su pauta aprobada reduce significativamente, en comparación con placebo, la tasa mensual de crisis de AEH confirmadas por el investigador: al final del periodo se verificó una tasa de 1,11 crisis mensuales con el fármaco y de 2,18 con placebo, lo que suponía una reducción del 49 % (p= 0,003). Entre las variables secundarias, la más relevante fue la proporción de días con síntomas de angioedema en la parte 1, para la que no se alcanzó significación estadística en la comparación frente a placebo.
Con respecto a la seguridad, la evidencia deriva de hasta 381 pacientes con AEH que han recibido el fármaco a lo largo de su desarrollo clínico, si bien los más robustos son los del ensayo pivotal, donde hasta 108 pacientes llegaron a recibir al menos una dosis en su parte 2. El análisis combinado de los datos demostró que los eventos adversos son más frecuentes con berotralstat 150 mg/día que con placebo, destacando una mayor frecuencia de: nasofaringitis (28 % vs. 23 %), cefalea (14 % vs. 5 %), diarrea (13 % vs. 0 %), dolor abdominal (12 % vs. 5 %), infecciones del tracto respiratorio superior (10 % vs. 2,6 %), vómitos (8 % vs. 2,6 %), infecciones del tracto urinario (6 % vs. 0 %) y reflujo gastroesofágico (5 % vs. 0 %). Las alteraciones bioquímicas más comúnmente observadas fueron la elevación de los niveles de transaminasas hepáticas, y se notificó hipersensibilidad/rash en el 3 % de los pacientes tratados con berotralstat.
No obstante, de los eventos adversos que se consideraron relacionados con el tratamiento (21 %), la práctica totalidad fueron leves-moderados en severidad. Por ejemplo, los episodios gastrointestinales, de los más comunes con el tratamiento, se registraron mayoritariamente en los 3 primeros meses de tratamiento y se resolvieron en unos 3-4 días sin farmacoterapia específica y sin necesidad de interrumpir la pauta profiláctica. Los eventos considerados graves, ninguno de ellos relacionado con el tratamiento, también fueron más frecuentes con berotralstat (10 % vs. 5 % con placebo), siendo la crisis de AEH el más común (3,5 %). En cualquier caso, no se registró ninguna muerte en los estudios y la discontinuación de la profilaxis por motivos de seguridad no fue muy frecuente (8 % vs. 3 % con placebo). Respecto a grupos etarios, el perfil de seguridad descrito en pacientes adolescentes es similar al observado en adultos, pero los pacientes más mayores reportaron mayor incidencia de alteraciones hepáticas potencialmente relacionadas con el fármaco, efectos adversos gastrointestinales y mayor incidencia de anormalidades de laboratorio más relevantes (AEMPS, 2023).
Aspectos innovadores
Berotralstat es un nuevo fármaco de molécula pequeña, activo por vía oral, específicamente diseñado para actuar como un inhibidor potente y selectivo de la actividad serina proteasa de la calicreína plasmática, una enzima que fisiológicamente participa en la activación del complemento mediante la escisión del cininógeno de alto peso molecular para liberar bradicinina. Dado que la bradicinina es un potente vasodilatador que aumenta la permeabilidad vascular y desencadena las crisis de edema, se comprende que el nuevo fármaco, capaz de suprimir la producción de bradicinina en células endoteliales, puede tener potencial terapéutico en pacientes con deficiencia o disfunción de la proteína inhibidora de la C1-esterasa (INH-C1), en quienes está patológicamente aumentada la actividad normal de la calicreína plasmática y la liberación de bradicinina. Así, el medicamento ha sido autorizado para la prevención rutinaria –terapia oral diaria– de las crisis recurrentes de angioedema hereditario (AEH) en pacientes adultos y adolescentes de al menos 12 años de edad, no pudiéndose usar para el tratamiento de las crisis agudas.
En ese supuesto terapéutico de profilaxis, hasta ahora se disponía de otros tratamientos eficaces: los inhibidores de C1 esterasa de origen plasmático (de administración intravenosa o subcutánea) y el anticuerpo monoclonal lanadelumab (subcutáneo). Las principales guías de práctica clínica y las recomendaciones de expertos coinciden en que estas opciones son utilizadas habitualmente en la práctica clínica de forma indistinta.
La aprobación de berotralstat en la pauta de 150 mg/día se sustentó en los resultados de un estudio pivotal de fase 3 y adecuado diseño (aleatorizado, multicéntrico, doble ciego, controlado con placebo, de 3 grupos paralelos y diferenciado en 3 partes), que incluyó a 121 pacientes con AEH de tipo I y II. En la parte 1, se demostró que un tratamiento durante 6 meses con el fármaco reducía en un 44 % la tasa mensual de crisis de AEH confirmadas por el investigador (variable primaria) en comparación con placebo12, alcanzando significación estadística (p< 0,001). La eficacia del fármaco se observa desde el primer mes y se mantiene hasta el final de ese periodo, cuando casi un 60 % de los pacientes (vs. 25 % con placebo) ha reducido la frecuencia de crisis a más de la mitad respecto al inicio, con una reducción relativa del 49 % frente a placebo también en la incidencia de crisis que requieren tratamiento agudo. Hay que citar, no obstante, que se vio una diferencia por sexos en la eficacia, superior en hombres (reducción del 65 % de la tasa mensual de crisis frente a placebo vs. -29 % en mujeres), y que, aunque la tendencia es favorable, el fármaco no ha podido demostrar una mejora estadísticamente significativa frente a placebo en las escalas de calidad de vida según reporte de los pacientes. En la parte 2 del estudio se confirmó que la eficacia de berotralstat se mantiene a largo plazo, al menos en tratamientos de casi 1 año de duración (48 semanas): los pacientes tratados con el fármaco todo ese tiempo pasaron de una tasa basal de 3,1 crisis mensuales a 1,1, lo que supone una reducción del 67 %.
Un estudio de soporte de similar diseño13, también dividido en 3 partes y realizado en población japonesa (N= 19), aportó resultados que sustentan los divulgados para el pivotal: frente a placebo, un tratamiento con la pauta aprobada de berotralstat redujo en un 49 % la tasa de crisis mensuales de AEH (al final del periodo tenían 1,1 crisis mensuales vs. 2,2 con placebo).
Si bien los resultados de estas investigaciones se pueden considerar representativas y extrapolables a la práctica clínica14, la evidencia no está exenta de limitaciones, entre las que sobresale la ausencia de un comparador activo (como INH-C1 o lanadelumab), que habría sido más adecuada y deseable; la ausencia de significación estadística en la escala de calidad de vida o en la eficacia según localización anatómica de la crisis, que pondría en cuestión la relevancia clínica de la reducción de la tasa de crisis; o el pequeño número de pacientes adolescentes y la ausencia de pacientes mayores de 75 años o de menos de 40 kg de peso.
En términos de seguridad, el fármaco ha mostrado un perfil toxicológico relativamente benigno y bien tolerado. Aunque se relaciona con una incidencia de eventos adversos mayor que placebo, en menos de un cuarto de los pacientes se relacionan con el tratamiento. Entre las reacciones adversas más comunes a berotralstat destacan las de tipo digestivo (≈15 %; sobre todo dolor abdominal, diarrea y vómitos, en su mayoría pasajeras) así como infecciones del tracto respiratorio superior (incluida nasofaringitis), cefalea, alteraciones hepáticas e hipersensibilidad/rash. Pero en su práctica totalidad son leves-moderadas, en ningún caso fatales, y no se relacionan con una alta tasa de interrupciones del tratamiento. En todo caso, se requieren más datos con su uso a largo plazo para esclarecer el perfil beneficio-riesgo en profilaxis prolongada en el contexto de una patología crónica.
El posicionamiento de berotralstat en el arsenal terapéutico se complica por la ausencia de comparaciones directas con las otras alternativas aprobadas en su indicación. Si se recurre a comparaciones indirectas, de robustez estadística que limita la extracción de conclusiones (por las diferencias metodológicas y de población incluida), se puede aludir a un meta-análisis en red recientemente publicado que ha comparado la efectividad del nuevo fármaco con la de lanadelumab basándose en los resultados de estudios de fase 3. Sus hallazgos apuntan a que lanadelumab es significativamente más efectivo que berotralstat en cuanto a la tasa de crisis mensuales de AEH y a la proporción de pacientes con reducción de ≥ 90 % en las crisis agudas (Watt et al., 2023). De hecho, en el ensayo pivotal de lanadelumab se vio que su pauta subcutánea bisemanal de 300 mg redujo en hasta un 87% la frecuencia de crisis frente a placebo tras 6 meses (Fernández-Moriano, 2021).
En definitiva, con un mecanismo de acción prácticamente idéntico a lanadelumab, con el que comparte indicación, berotralstat puede ser un tratamiento eficaz y bien tolerado en la prevención de las crisis de angioedema a largo plazo, mejor que la ausencia de tratamiento preventivo. Parece que va posicionarse como una opción terapéutica más, alternativa a los tratamientos ya disponibles (sobre todo, en pacientes que no toleren o no estén adecuadamente protegidos con otro fármaco o no hayan sido controlados satisfactoriamente con terapia aguda repetida15), respecto a los cuales no parece demostrar ningún beneficio incremental salvo la diferencia de la vía oral (frente a la intravenosa o subcutánea de sus alternativas), que podría impactar potencialmente en una mejor conveniencia para el paciente, aunque no se ha estudiado si mejora la adherencia al tratamiento. La principal guía de práctica clínica europea de la enfermedad –WAO/EEAIC 2022 (Maurer et al., 2022)– recomienda lanadelumab, los inhibidores de C1 y berotralstat como tratamiento de primera línea para la profilaxis a largo plazo de los pacientes con AEH tipo I o II, de lo que se deduce que el nuevo fármaco no va a suponer una revolución de la terapéutica estándar.