Los medicamentos de terapia avanzada (MTA o Advanced Therapy Medicinal Products, ATMP) ofrecen nuevos e innovadores tratamientos para las enfermedades. Están basados en la terapia génica, la terapia celular somática o la ingeniería tisular. El marco legal para las ATMP en la Unión Europea está establecido en la Regulation (EC) No 1394/2007 on advanced therapy medicinal products que asegura el libre movimiento de estas medicinas dentro de la Unión Europea y el acceso a los mercados. La regulación (EC) nº 1394/2007 también establece el nuevo Comité en Terapias avanzadas (CAT) cuya responsabilidad fundamental consiste en preparar un proyecto de opinión sobre cada nueva solicitud de medicamento de terapia avanzada planteada a la Agencia Europea de Medicamentos, antes de que el Comité de Medicamentos de Uso Humano (CHMP, Committee for Medicinal Products for Human Use) de la misma adopte una opinión definitiva sobre la concesión, modificación, suspensión o revocación de una autorización de comercialización para el medicamento en cuestión.
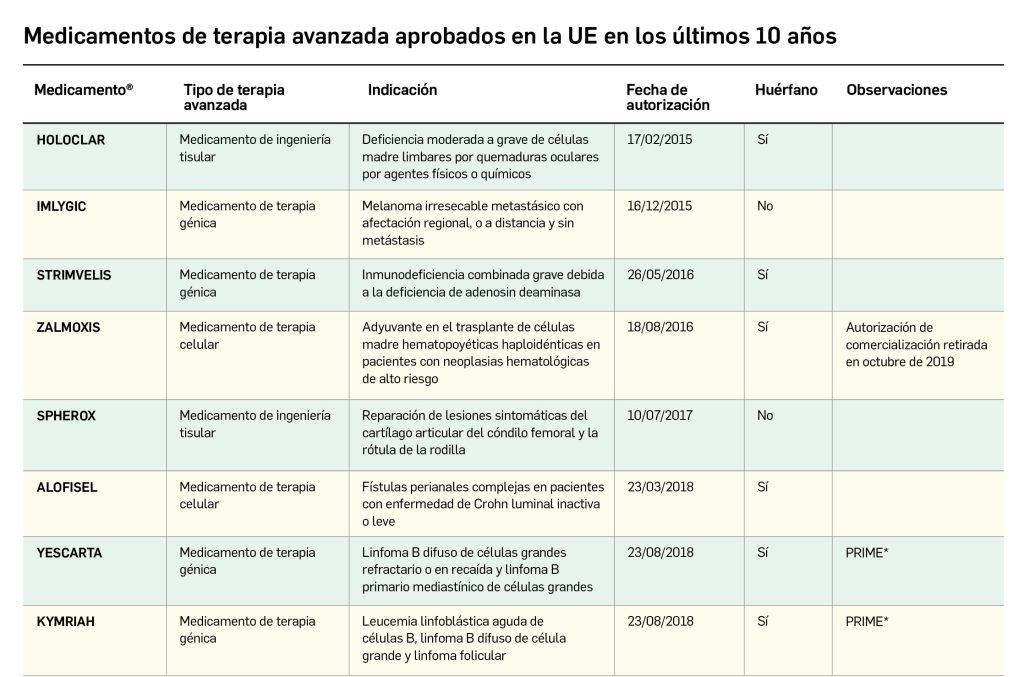
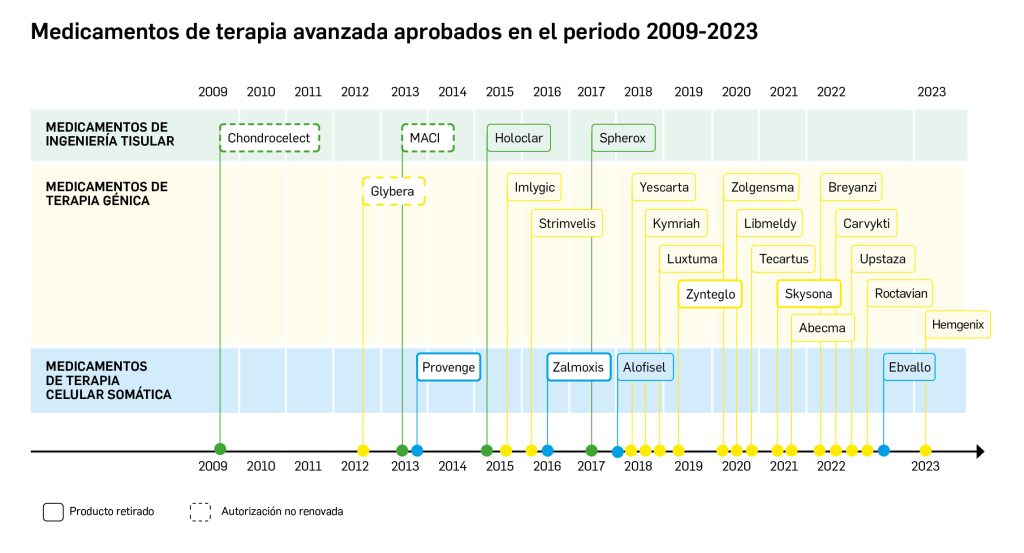
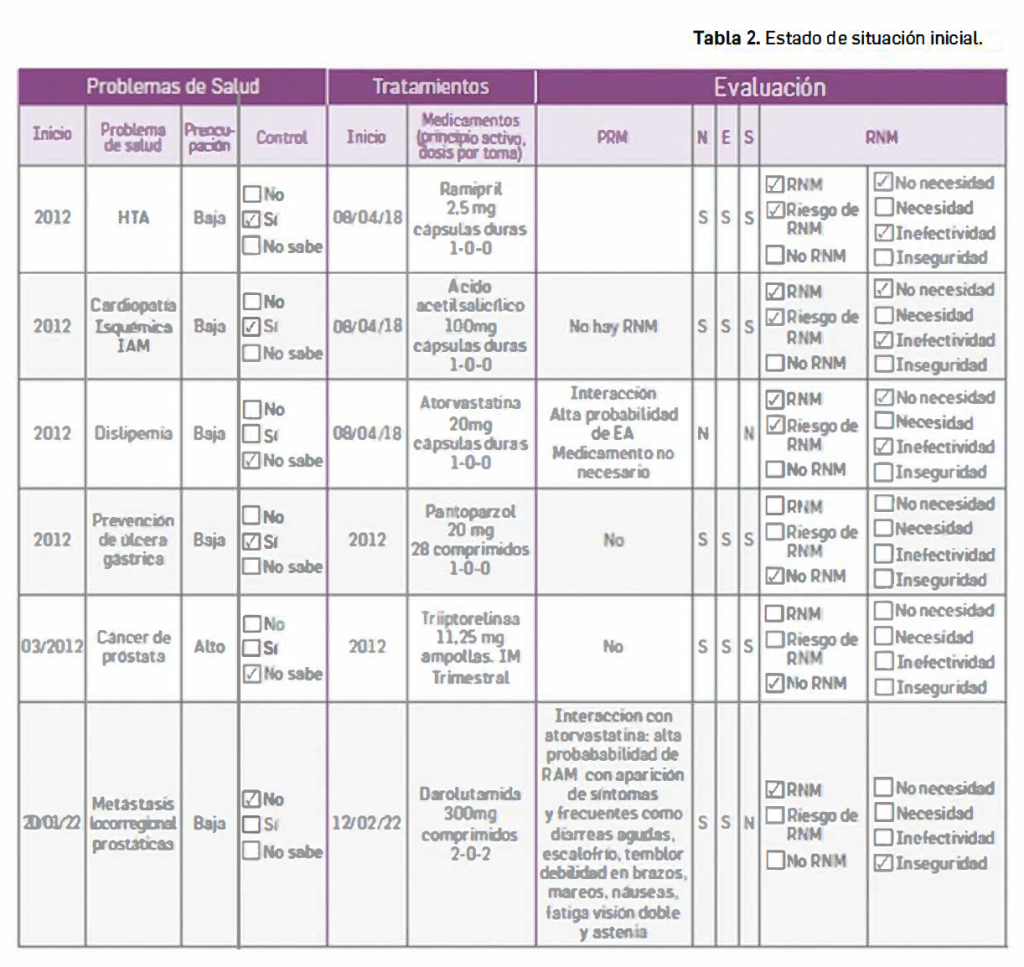
La siguiente tabla recoge los medicamentos de terapia avanzada que han recibido autorización de comercialización en la UE durante en los últimos 10 años (2013-2023).