Resumen
La nueva vacuna tetravalente del dengue es una vacuna de virus vivos atenuados cuyo mecanismo de acción consiste en la estimulación de la producción de anticuerpos de larga duración que confieren protección frente a los cuatro serotipos conocidos del virus del dengue como consecuencia de la replicación viral que se produce tras la vacunación. El medicamento es capaz de activar distintos tipos de mecanismos inmunitarios, incluyendo la producción de anticuerpos de unión, activación anticuerpo-dependiente del sistema del complemento, producción de anticuerpos frente a la proteína no estructural 1 (NS1) y la proliferación de linfocitos T y células NK; en base a ello, ha sido autorizado con indicación en la prevención del dengue en personas a partir de los 4 años de edad, siempre que se sigan las recomendaciones oficiales de vacunación.
La eficacia clínica de la vacuna fue evaluada en un estudio pivotal de fase 3 doble ciego, aleatorizado y controlado con placebo en participantes sanos de entre 4 y 16 años residentes en zonas endémicas de dengue, con el objetivo principal de determinar la disminución del riesgo de enfermedad a partir de los 30 días siguientes a la administración de la segunda dosis hasta pasados 12 meses. Los resultados publicados respaldan el cumplimiento del objetivo primario del estudio: se estimó una eficacia del 80,2% (IC95%: 73,3-85,3%; p< 0,001), ampliamente por encima del límite preestablecido del 25% en el extremo inferior del intervalo. También se cumplió el principal objetivo secundario: se observó una eficacia estimada del 90,4% en la prevención de la hospitalización en los 18 meses posteriores a la segunda dosis. Aunque había cierta tendencia al descenso de la eficacia vacunal con el paso del tiempo, una dosis de refuerzo a los 36 meses supuso un nuevo incremento de la protección. El perfil de seguridad de esta vacuna parece benigno y los eventos adversos asociados a su administración son por lo general leves o moderados y pasajeros, sin que se hayan identificado riesgos preocupantes. Los eventos más frecuentes asociados a la recepción de la vacuna en los estudios controlados con placebo fueron dolor en el sitio de inyección (42%) dolor de cabeza (34%), mialgia (28%) y malestar general (23%). En el caso de la población pediátrica de > 4 años, se observaron fiebre, infecciones del tracto respiratorio superior, nasofaringitis, faringoamigdalitis y enfermedad gripal en mayor proporción que entre los adultos. No se ha podido excluir la posibilidad de dengue grave en individuos seronegativos al infectarse por los serotipos 3 o 4 tras recibir la vacuna.
A pesar de no tratarse de la primera vacuna tetravalente de virus vivos atenuados frente al dengue –pues Dengvaxia® fue autorizada por la Comisión Europea en 2018–, Qdenga® parece superar las limitaciones de su predecesora, con una capacidad de prevención elevada frente a todos los serotipos del virus –aunque probablemente mayor frente a DENV-2– y un perfil de seguridad benigno, en el que no se ha identificado hasta ahora un mayor riesgo de dengue grave entre las personas vacunadas e inicialmente seronegativas, aunque esto deberá ser confirmado en análisis basados en datos del mundo real, con un número de vacunados muy superior al de los estudios clínicos. Por tanto, teniendo en cuenta las limitaciones relativas a la prevención basada en evitar en contacto y a la ausencia de otras medidas farmacológicas de prevención o tratamiento con antivirales, la nueva vacuna tetravalente supone un significativo avance en la lucha frente a una infección actualmente en expansión y de consecuencias potencialmente muy graves.
Aspectos fisiopatológicos
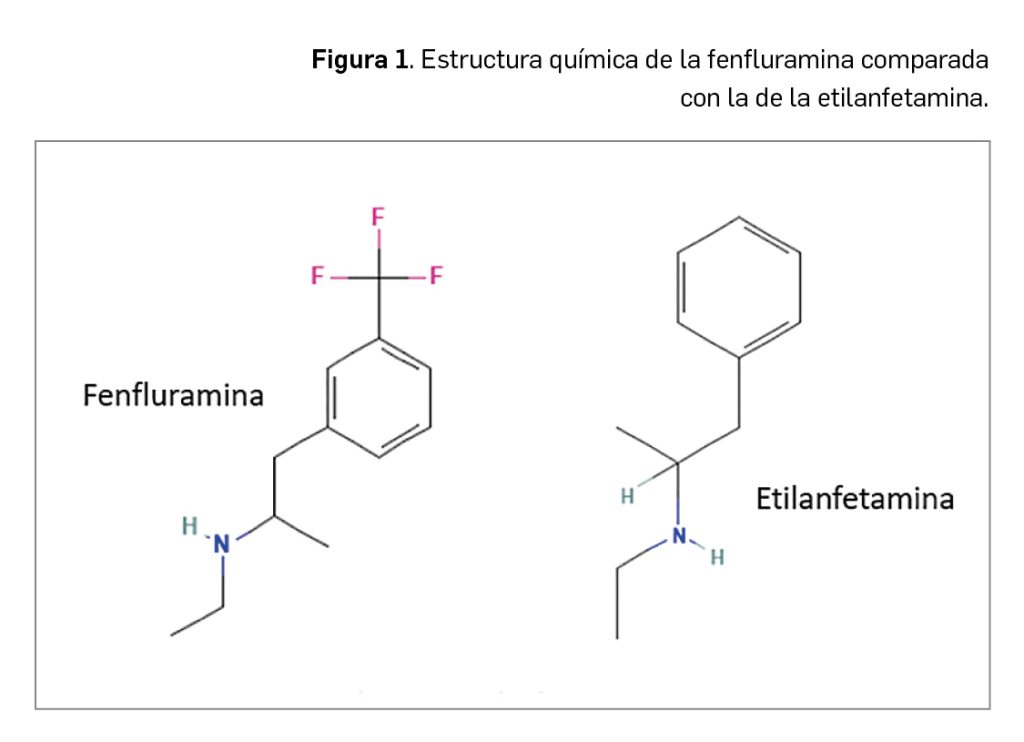
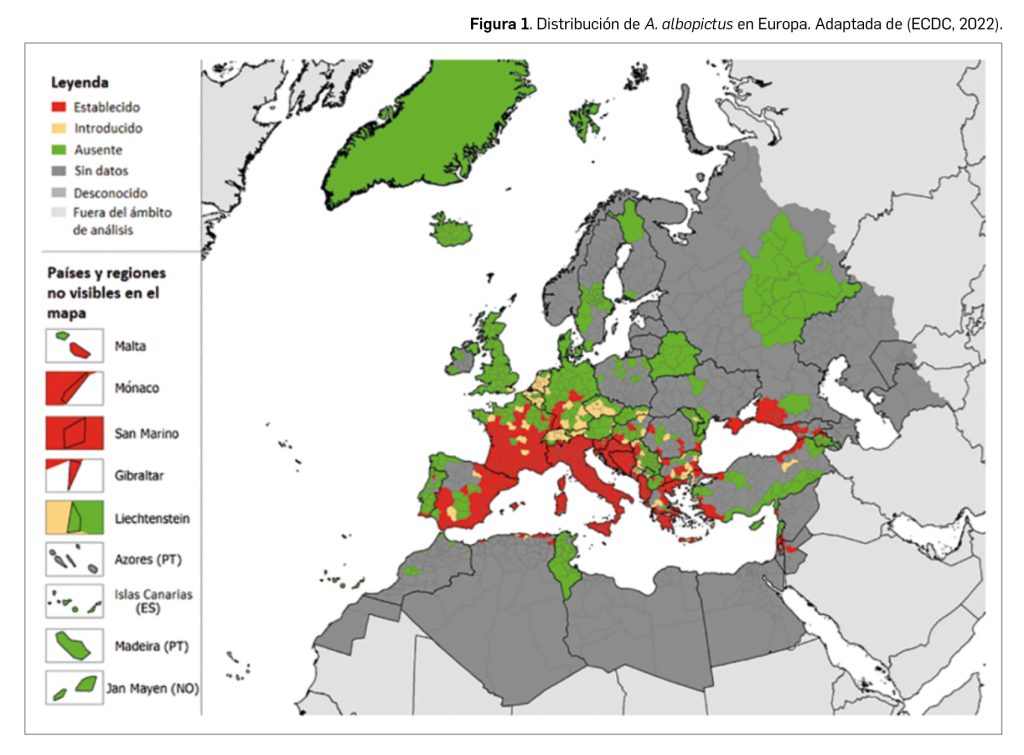
El virus del dengue se clasifica dentro de la familia Flaviviridae, en el género Flavivirus. Aunque es habitual referirse al virus del dengue de forma singular, se distinguen al menos cuatro serotipos distintos del virus, denominados por lo general como DENV-1, DENV-2, DENV-3 y DENV-4, todos ellos patogénicos y capaces de ocasionar desde una infección asintomática hasta enfermedad grave. La trasmisión a humanos se produce a través de la picadura de distintas especies de mosquitos del género Aedes (A. albopictus y A. aegypti)1, cuya distribución se ha ido expandiendo desde zonas subtropicales y tropicales hacia regiones de clima moderado en todo el mundo (Figura 1). Se estima que se producen entre unos 300 y 500 millones de infecciones cada año, de las cuales unos 100 millones provocan algún síntoma (Bhatt et al., 2013), con 2500 millones de personas en riesgo de infección. Las áreas consideradas endémicas son principalmente las zonas tropicales y subtropicales: América Latina –incluyendo México, pero con baja o nula presencia en Chile y Argentina– y el Caribe, sudeste y sur de Asia y buena parte del África subsahariana. Se debe tener en cuenta que A. albopictus está presente en amplias zonas del continente europeo y que en esta región ya se han reportado algunos casos autóctonos2, aunque la mayor parte de los casos registrados en Europa son adquiridos e importados desde zonas endémicas.
Tras la entrada del virus al torrente sanguíneo como consecuencia de la picadura del mosquito se produce su introducción en las células, un proceso mediado por la glicoproteína de la envoltura (E) viral. La infección de las células desencadena una respuesta inmunitaria, tanto innata como adaptativa: por un lado, se activa la producción de interferones antivirales (como el IFN-α); sin embargo, el virus del dengue tiene la capacidad de inhibir la producción de interferones y su actividad antiviral (Jones et al., 2005). Además, la producción de anticuerpos frente a antígenos virales (como la proteína E o la proteína precursora de la membrana) media la lisis y la citotoxicidad celular y bloquea la capacidad del virus para penetrar en las células. Estos anticuerpos producen una inmunidad de larga duración frente a la infección posterior por un virus del mismo serotipo, pero no confieren protección frente al resto.
Aunque los niños de corta edad, especialmente los menores de un año, son el principal grupo de riesgo de dengue grave, en la población infantil la enfermedad es con frecuencia asintomática o se manifiesta con escasos síntomas. En cambio, en los adultos es más frecuente que se presente de forma sintomática. Los síntomas suelen ocurrir tras entre 4 y 7 días desde la picadura del mosquito y comienzan con una fase febril, caracterizada por fiebre alta (> 38,5ºC), con dolor de cabeza, vómitos y mialgias. En esta fase se pueden presentar hemorragias (en la mucosa gastrointestinal, por ejemplo) o signos de fragilidad capilar, como petequias. La fase febril puede progresar a una fase crítica de entre 1 y 2 días de duración, especialmente en niños menores de un año o en pacientes inmunocomprometidos. Durante esta fase también pueden ocurrir manifestaciones hemorrágicas y un síndrome de extravasación vascular que puede conducir a sangrados, shock y fallo orgánico. Finalmente, se puede distinguir una fase de recuperación que dura entre 2 y 4 días en la que se resuelven la extravasación y las hemorragias, aunque otros síntomas, como la fatiga extrema, pueden mantenerse durante varias semanas.
Se asume que buena parte de los síntomas del dengue y, especialmente, del dengue grave, están relacionados con la respuesta que se desencadena en el huésped tras la infección. En este sentido, la gravedad de la enfermedad se ha vinculado con determinadas variantes genéticas –por ejemplo, las variantes alélicas del complejo mayor de histocompatibilidad HLA-AA24 o HLA-AB513– y también con la activación del sistema inmunitario a través de la función de los linfocitos y de la producción de citocinas proinflamatorias y de anticuerpos. De hecho, la fiebre hemorrágica, una de las complicaciones más graves de la infección y que está relacionada con la permeabilidad capilar, se produce en buena parte de los casos con una viremia baja o incluso indetectable (Murugesan et al., 2020).
Aunque la infección puede ser asintomática, en la mayor parte de los casos se producirán al menos algunos síntomas que, junto con otros factores, entre los que destaca el haber estado en una zona endémica de dengue, orientarán el diagnóstico. En este sentido, la Organización Mundial de la Salud (OMS), elaboró en el año 2009 una clasificación que establece tres categorías de gravedad de la infección por el virus del dengue.

No existe por ahora ningún antiviral específico para tratar la infección, por lo que el tratamiento estándar es de soporte en función de los síntomas presentes. Los casos leves se pueden abordar a nivel domiciliario con antipiréticos y analgésicos –paracetamol o AINEs, principalmente– y una correcta hidratación. En pacientes que requieren de hospitalización, a estas medidas se añadirán, en su caso, las necesarias para abordar la extravasación plasmática –que pueden ir desde la rehidratación en casos leves hasta la transfusión de sangre en casos graves–, el shock hipovolémico o las hemorragias.
La prevención de la infección comprende tanto las medidas orientadas a evitar el contacto con el mosquito como la vacunación. Entre las primeras, el uso de repelentes en zonas endémicas puede reducir el riesgo de picadura, mientras que también se han implementado medidas públicas de control del mosquito como el uso de insecticidas en espacios públicos, cuya eficacia se ve limitada porque en estas zonas es común que los mosquitos desarrollen su ciclo vital dentro de los hogares. A las personas que viajan a zonas endémicas se les recomienda el uso de repelentes y de prendas que cubran la mayor cantidad de superficie corporal posible mientras se encuentren al aire libre. En interiores, el uso de aire acondicionado y mantener ventanas y puertas exteriores cerradas pueden reducir el riesgo de contacto con los mosquitos.
Por otro lado, en 2018 la EMA autorizó el uso de la primera vacuna frente al dengue (Dengvaxia®) con indicación en la prevención de la enfermedad provocada por cualquiera de los 4 serotipos conocidos del virus (vacuna tetravalente) en personas de entre 6 y 45 años con infección confirmada mediante un test diagnóstico. Se trata de una vacuna de virus vivos atenuados y que, de acuerdo a los resultados de los estudios clínicos que condujeron a su aprobación, presenta una eficacia de entre el 80% y el 95% en la prevención de dengue grave (asociado a fiebre hemorrágica o necesidad de hospitalización).
A pesar de estar autorizada en la Unión Europea, esta vacuna nunca ha llegado a comercializarse en España. Con posterioridad a su autorización, se han observado determinados problemas de eficacia y algunos aspectos relativos a su seguridad que han limitado su utilización. Concretamente, la eficacia vacunal en personas seronegativas es baja y, además, en estos casos la vacunación parece incrementar el riesgo de dengue grave, algo que no ocurre en personas seropositivas antes de la vacunación. Por tanto, teniendo en cuenta la tendencia expansiva que presenta actualmente el vector del dengue y la alta prioridad concedida por la Organización Mundial de la Salud a la estrategia de prevención basada en la vacunación frente al dengue, se entiende la importancia de disponer de una vacuna con una alta eficacia y con un adecuado perfil de seguridad con independencia del estado serológico previo.
Acción y mecanismo
La nueva vacuna tetravalente del dengue es una vacuna de virus vivos atenuados cuyo mecanismo de acción consiste en la estimulación de la producción de anticuerpos de larga duración que confieren protección frente a los cuatro serotipos conocidos del virus del dengue como consecuencia de la replicación viral que se produce tras la vacunación. La vacuna es capaz de activar distintos tipos de mecanismos inmunitarios, incluyendo la producción de anticuerpos de unión, activación anticuerpo-dependiente del sistema del complemento, producción de anticuerpos frente a la proteína no estructural 1 (NS1) y la proliferación de linfocitos T y células NK. En base a este mecanismo, el medicamento ha sido autorizado en su pauta de dos dosis subcutáneas (0 y 3 meses) con indicación en la prevención del dengue en personas a partir de los 4 años de edad.
La inmunogenicidad de la vacuna fue testada en distintos modelos animales (ratón y macaco) durante el desarrollo preclínico, pudiéndose comprobar tanto la producción de anticuerpos neutralizantes tras la administración de una primera dosis y de una dosis de refuerzo, como la protección frente a la enfermedad provocada por cualquiera de los serotipos del virus (EMA, 2022).
Aspectos moleculares
La vacuna compuesta por los cuatro serotipos vivos y atenuados del virus del dengue, que expresan tanto el antígeno precursor de la membrana (prM) como el antígeno de la envoltura (E) de cada uno de los serotipos. Todos tienen en común el esqueleto genético del serotipo 2 del virus.
La atenuación viral se ha conseguido fundamentalmente a través de tres mutaciones clave: la primera de ellas, en la región 5’ no codificante, en la que la citosina de la posición 57 se cambió por timina; la segunda se realizó sobre el gen que codifica para la proteína no estructural 1 (NS1) en la posición 2579, con un cambio de guanina por adenina, que produce un cambio en la traducción del codón del aminoácido glicina por ácido aspártico; por último, se realizó un cambio en el gen de la proteína no estructural 3 (NS3) en la posición 5270 al sustituir una adenina por una timina, lo que resulta en la sustitución en la traducción de un residuo de ácido glutámico por uno de valina.
Eficacia y seguridad clínicas
La eficacia y la seguridad clínicas de la vacuna tetravalente del dengue han sido adecuadamente evaluadas en un ensayo pivotal de fase 3 doble ciego, aleatorizado y controlado con placebo en participantes sanos de entre 4 y 16 años residentes en zonas endémicas de dengue4. Un total de 20 099 sujetos fueron aleatorizados en proporción 2:1 para recibir por vía subcutánea la vacuna (dos dosis separadas 90 días y una posterior dosis de refuerzo a los 48-54 meses de la primera dosis) o bien un placebo equivalente.
El estudio se estructuró en 5 partes: la parte 1, que sirvió para el análisis de la variable primaria de eficacia, comenzó en el día de vacunación y se alargó hasta la confirmación de 120 casos de dengue tras un seguimiento mínimo de 12 meses desde la segunda dosis; la parte 2 consistió en un periodo de 6 meses para el análisis de las variables secundarias de eficacia; la parte 3 permitió analizar la eficacia a largo plazo (con una duración de entre 2,5 y 3 años); y las partes 4 y 5 se refieren al periodo tras la dosis de refuerzo, con una duración de al menos 25 meses adicionales. La variable primaria de eficacia se definió como la disminución del riesgo de enfermedad (fiebre por dengue) a partir de los 30 días siguientes a la administración de la 2ª dosis hasta el final de la parte 15, mientras que la variable secundaria más relevante fue la prevención de la hospitalización desde los 30 días siguientes a la 2ª dosis hasta el final de la parte 2. Las características sociodemográficas estuvieron adecuadamente balanceadas entre los dos brazos de tratamiento, destacando el hecho de que el 72,3% de los participantes eran seropositivos para al menos un serotipo de dengue.
Según los resultados de eficacia publicados (Biswal et al., 2020), se alcanzó el objetivo principal: 12 meses después de la segunda dosis la proporción de participantes con fiebre por dengue fue del 0,5% en el grupo que recibió la vacuna vs. 2,4% en el brazo de placebo, estimándose una eficacia vacunal del 80,2% (IC95% 73,3-85,3%; p< 0,001). El extremo inferior del IC95% bilateral fue superior al 25%, de modo que se cumplió el criterio de la variable primaria de eficacia. Con respecto al principal objetivo secundario, la tasa de hospitalización por dengue al final de la parte 2 del estudio fue significativamente inferior en el grupo de vacunados (0,1% vs. 1,0%), con una eficacia estimada para la vacuna del 90,4% en la prevención de los ingresos (IC95% 82,6-94,7%; p< 0,001) que también cumplió el criterio predeterminado de al menos un 0% en el extremo inferior del IC95% bilateral.
El análisis de la eficacia a largo plazo (hasta 54 meses después de la segunda dosis) indica una protección que tiende a decaer con el tiempo: desciende hasta el 76,2% (IC95% 50,8-88,4) en la prevención de la hospitalización y al 56,2% (IC95%: 42,3-66,8) respecto a la prevención de la fiebre a los 24 meses de la segunda dosis, y hasta el 70,8% (IC95% 49,6-83,0) y el 45,0% (IC95% 32,9-55,0), respectivamente, a los 36 meses (Rivera et al., 2022). A partir de este punto se planificó la administración de una dosis de refuerzo, de modo que la eficacia vacunal a los 48 meses aumentó hasta el 96,4% (IC95% 72,2-99,5) para prevenir la hospitalización y hasta el 62,8% (IC95% 41,4-76,4) en la prevención de la fiebre (AEMPS, 2022).
Cabe mencionar dos aspectos importantes en relación con la eficacia de la nueva vacuna. Por un lado, que la capacidad de prevención de la hospitalización y la fiebre, aunque elevada, es variable dependiendo del serotipo, resultando superior frente a la infección por DENV-2. Además, la inmunoprotección también fue superior en aquellos participantes que eran seropositivos al inicio del estudio (Tabla 2).
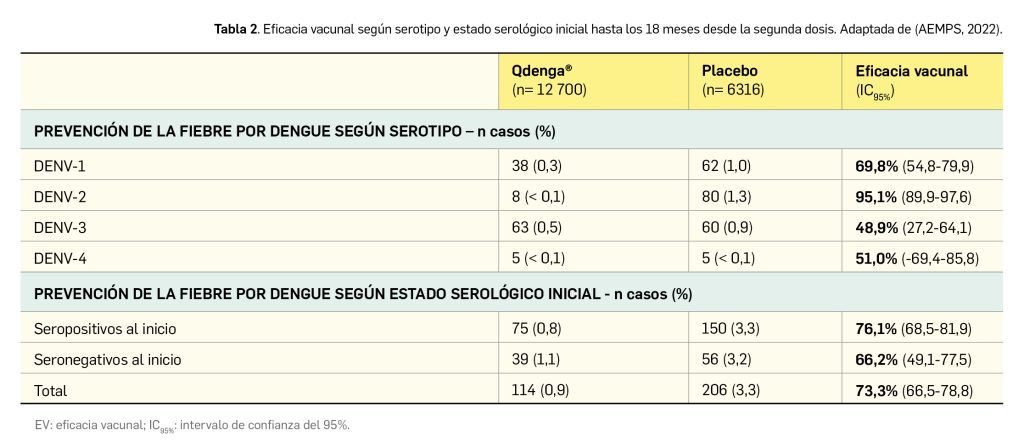
En este sentido, los títulos detectados de anticuerpos un mes después de la segunda dosis fueron superiores en los participantes inicialmente seropositivos (MGT6 mínima: 1129,4 frente a DENV-4; MGT máxima: 4897,4 frente a DENV-2). Entre los individuos seronegativos en el periodo basal, el título de anticuerpos también fue superior frente al serotipo 2 del virus pasado un mes desde la segunda dosis (MGT mínima: 143,9 frente a DENV-4; MGT máxima: 1729,9 frente a DENV-2).
En términos de seguridad se dispone de datos procedentes de 18 estudios clínicos con un total de 27 573 participantes que recibieron vacuna, siendo los más robustos los que proceden de ensayos aleatorizados y controlados con placebo (incluido el pivotal); se tienen datos consolidados para un subconjunto de 5555 participantes (3830 recibieron la vacuna y 1725, placebo).
Si se consideran los eventos adversos reportados en respuesta a una solicitud concreta del investigador7, la mayor parte fueron de intensidad leve o moderada, con un 1,3% de eventos locales y un 4,1% de eventos sistémicos de grado 3. Los más frecuentes (> 20%) fueron: dolor en el sitio de inyección (41,8% con la vacuna vs. 25,4% con placebo), dolor de cabeza (33,8% vs. 30,1%), mialgia (28,0% vs. 20,5%) y malestar general (22,9% vs. 20,7%). La incidencia de eventos adversos reportados de manera espontánea fue similar en el grupo de la vacuna (21,3%) y de placebo (22,8%), considerados en su mayoría como no relacionados con el fármaco en estudio. Entre los que sí se relacionaron con el medicamento, la incidencia fue algo superior en el grupo de la vacuna (3,0% vs. 1,7%), en especial para los casos de nasofaringitis (2,6%). Aunque con una incidencia baja, en el estudio pivotal se observó una mayor proporción de casos de dengue grave entre los participantes que recibieron la vacuna hasta el tercer año desde la segunda dosis (0,13% vs. 0,05%).
No parece haber diferencias sustanciales en el perfil de reacciones adversas en la población pediátrica (4 a 17 años) en comparación con los adultos. De acuerdo a los datos recogidos en la ficha técnica, los eventos adversos informados con mayor frecuencia en población pediátrica fueron fiebre (11% vs. 3% en adultos), infección del tracto respiratorio superior (11% vs. 3%), nasofaringitis (6 % vs. 0,6 %), faringoamigdalitis (2% vs. 0,3%) y enfermedad de tipo gripal (1% vs. 0,1%). En cambio, el eritema en el lugar de inyección fue más frecuente en adultos (27% vs. 2%).
Aspectos innovadores
El nuevo medicamento es una vacuna de virus vivos atenuados cuyo mecanismo de acción consiste en la estimulación de la producción de anticuerpos de larga duración que confieren protección frente a los cuatro serotipos conocidos del virus del dengue como consecuencia de la replicación viral que se produce tras la vacunación. La vacuna es capaz de activar distintos tipos de mecanismos inmunitarios, incluyendo la producción de anticuerpos de unión, activación anticuerpo-dependiente del sistema del complemento, producción de anticuerpos frente a la proteína no estructural 1 (NS1) y la proliferación de linfocitos T y células NK. En base a este mecanismo, ha sido autorizada con indicación en la prevención del dengue en personas a partir de los 4 años de edad, debiéndose seguir las recomendaciones oficiales para su uso.
La eficacia clínica de la vacuna tetravalente del dengue se ha examinado en un estudio pivotal de fase 3 doble ciego, aleatorizado y controlado con placebo en participantes sanos de entre 4 y 16 años (N: 20 099) residentes en zonas endémicas de dengue. Los resultados revelan que se cumplió el objetivo principal: se estimó una eficacia vacunal –en términos de disminución del riesgo de enfermedad a partir de los 30 días siguientes a la administración de la segunda dosis hasta el final de la parte 1 del estudio– del 80% (p< 0,001), ampliamente por encima del límite preestablecido de significación estadística. Entre las variables secundarias, destaca una eficacia estimada del 90% en la prevención de la hospitalización tras 18 meses desde la segunda dosis. Si bien la eficacia vacunal parece descender con el tiempo (hasta el 45% en la prevención de fiebre a los 36 meses desde la 2ª dosis y hasta el 71% en la prevención de la hospitalización por dengue), la revacunación permite recuperar una elevada protección (esos valores crecen hasta el 63% y el 96%, respectivamente).
De manera consistente, se ha observado a lo largo del estudio una mayor eficacia vacunal en los participantes que eran inicialmente seropositivos para el serotipo 2 del virus (DENV-2), unos resultados coherentes con la mejor respuesta inmunitaria observada en estos individuos en términos de presencia de anticuerpos neutralizantes frente al virus (títulos hasta 10 veces superiores que frente al resto de serotipos). El perfil de seguridad de la vacuna parece benigno y los eventos adversos asociados a la administración son por lo general leves o moderados y pasajeros. Los eventos más frecuentes entre quienes recibieron la vacuna en los estudios clínicos aleatorizados y controlados con placebo fueron dolor en el sitio de inyección (42%) dolor de cabeza (34%), mialgia (28%) y malestar general (23%). Entre la población pediátrica (> 4 años) se observó una incidencia mayor que en adultos de fiebre, infecciones del tracto respiratorio superior y enfermedad gripal. Pese a que en el análisis de seguridad no se ha hallado ningún riesgo importante para la vacuna, en el EPAR se mencionan como riesgos potenciales las reacciones anafilácticas, la probabilidad de sufrir dengue debido a una pérdida de la eficacia con el tiempo y el dengue grave por los serotipos 3 y 4 en participantes seronegativos. Además, no se conoce su tolerabilidad en mujeres embarazadas, debido a que el embarazo y la lactancia fueron criterios de exclusión en los distintos estudios.
Esta no es la primera vacuna tetravalente de virus vivos atenuados autorizada en la Unión Europea frente al dengue, pues Dengvaxia®, autorizada en 2018, era del mismo tipo. Sin embargo, los problemas de eficacia y seguridad de esta vacuna en individuos seronegativos comprometieron seriamente su utilización. A tenor de lo expuesto, Qdenga® parece superar esta limitación de su predecesora, con una capacidad de prevención elevada en individuos tanto seropositivos como seronegativos, frente a todos los serotipos del virus –probablemente mayor frente a DENV-2– y un perfil de seguridad benigno, en el que no se ha podido identificar hasta ahora un mayor riesgo de dengue grave entre las personas vacunadas e inicialmente seronegativas, aunque esto deberá ser confirmado en análisis basados en datos del mundo real, con un número de vacunados muy superior al de los estudios clínicos. Por tanto, teniendo en cuenta las limitaciones relativas a la prevención basada en evitar en contacto y a la ausencia de otras medidas farmacológicas de prevención o tratamiento con antivirales, la nueva vacuna tetravalente supone un significativo avance en la lucha frente a una infección actualmente en expansión y de consecuencias potencialmente muy graves.