Resumen
Anifrolumab es un nuevo anticuerpo monoclonal que se une con alta afinidad y especificidad a la subunidad 1 del receptor del interferón de tipo 1 (IFNAR1). En condiciones fisiológicas, la unión del interferón de tipo 1 (IFN-1) a este receptor activa la cascada de señalización JAK-STAT, que regula la expresión de los genes relacionados con los interferones. Esto permite reducir la producción de IFN-1, habitualmente sobreexpresado en pacientes con lupus eritematoso sistémico (LES) y que parece estar relacionado con la etiopatogenia debido a la estimulación que induce en la maduración de las células dendríticas, la producción de autoanticuerpos, la producción de inmunocomplejos o la inflamación orgánica. Anifrolumab ha sido autorizado con indicación en el tratamiento del LES con autoanticuerpos positivos, activo de moderado a grave en pacientes adultos, en combinación con el tratamiento estándar.
Su eficacia ha sido examinada principalmente en el estudio pivotal TULIP-2, un estudio de fase 3, aleatorizado, doble ciego, multicéntrico y controlado con placebo, en el que participaron pacientes adultos con LES crónico de moderado a grave a pesar de recibir el tratamiento estándar. La variable principal de eficacia en este estudio fue la tasa de pacientes con respuesta en la semana 52 según criterios BICLA, para la que anifrolumab fue superior a placebo (48% vs 32%; p= 0,0013). Respecto a las variables secundarias, la tasa de respuesta en la semana 52 fue también superior en el brazo de anifrolumab en el grupo de pacientes con alta expresión del gen de IFN (48% vs. 31%; p= 0,0022). Además, el 52% de los pacientes del brazo de anifrolumab vs. 30% del brazo de placebo pudieron reducir la dosis a ≤ 7,5 mg/día de prednisona o equivalente. Estos datos se ven apoyados por los resultados a largo plazo de un estudio de extensión en pacientes que completaron los estudios de fase 3, especialmente en la variable de reducción de corticoides, con un resultado favorable para anifrolumab tras un periodo de 4 años (29% con placebo vs. 10% en el grupo de anifrolumab requerían una dosis ≥ 7,5 mg/día de prednisona).
En relación al perfil de seguridad de anifrolumab, se dispone fundamentalmente de los datos de los estudios de fase 3 TULIP-1 y TULIP-2, con una exposición media de 761 días a anifrolu mab y de 487 días a placebo. La mayoría de participantes experimentó algún evento adverso, siendo más comunes en los grupos del nuevo fármaco respecto a placebo (88% vs. 81%). Los más frecuentes fueron nasofaringitis (18% vs. 11% con placebo), infecciones del tracto respiratorio superior (17% vs. 10%), bronquitis (11% vs. 5%) y herpes zóster (6% vs. 1%). En cambio, los eventos graves fueron más frecuentes en el brazo de placebo (16% vs. 11%).
En definitiva, anifrolumab incorpora un nuevo mecanismo de acción en el tratamiento del LES. Dada la escasez de tratamientos específicamente dirigidos a dianas relacionadas directamente con la patogenia de la enfermedad y teniendo en cuenta el alcance de los efectos adversos asociados al tratamiento crónico con corticosteroides e inmunosupresores, se puede considerar que, aunque de magnitud escasa, la eficacia de anifrolumab resultaría de relevancia clínica. La posibilidad de reducir la dosis de corticoides al introducir el fármaco podría explicar en parte la menor proporción de eventos adversos graves observados en los estudios de fase 3. Si bien sus efectos adversos a largo plazo no se conocen completamente y requieren de seguimiento dado su carácter inmunosupresor, los datos disponibles parecen apuntar a un perfil toxicológico más benigno que el asociado a las dosis altas de corticoides. Así, anifrolumab se posiciona por el momento como una alternativa a belimumab únicamente en pacientes adultos sin complicaciones renales o neuropsiquiátricas.
Aspectos fisiopatológicos
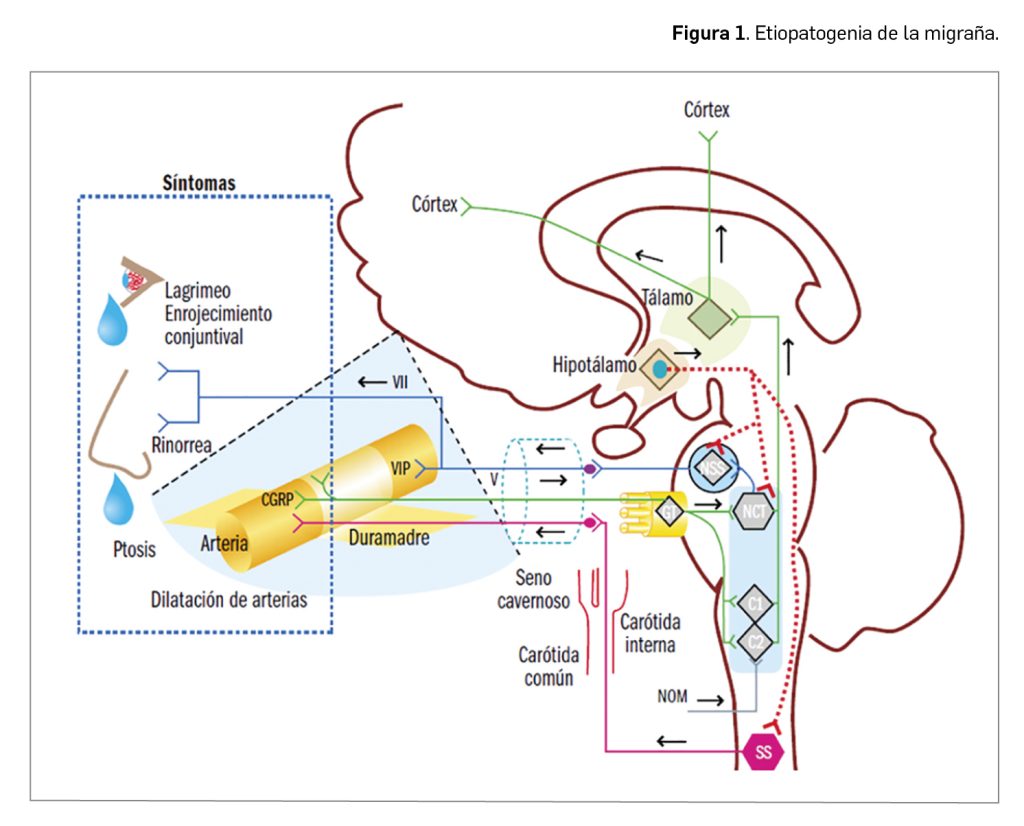
El lupus eritematoso sistémico (en adelante, LES) forma parte de un amplio conjunto de patologías, referidas habitualmente como enfermedades autoinmunes sistémicas, provocadas por una reacción anómala del sistema inmunitario del paciente frente a estructuras normales del propio paciente (autoinmune), capaces de afectar a varios órganos y/o tejidos (sistémico). En particular, el LES es un proceso muy heterogéneo en su presentación clínica (Figura 1): aunque las manifestaciones clínicas más frecuentes son las cutáneas, son también muy frecuentes la astenia y las artralgias. Además, puede presentarse artritis de forma intermitente, que afecta especialmente a las manos, pero también a otras articulaciones. La afectación visceral es frecuente y dispersa, pudiendo verse afectado cualquier órgano, desde el riñón, en forma de glomerulonefritis, hasta el sistema nervioso central (lupus neuropsiquiátrico) o los aparatos cardiovascular, pulmonar o digestivo. Es precisamente la diversidad de las posibles afectaciones orgánicas es la que determina la condición de sistémico del LES.
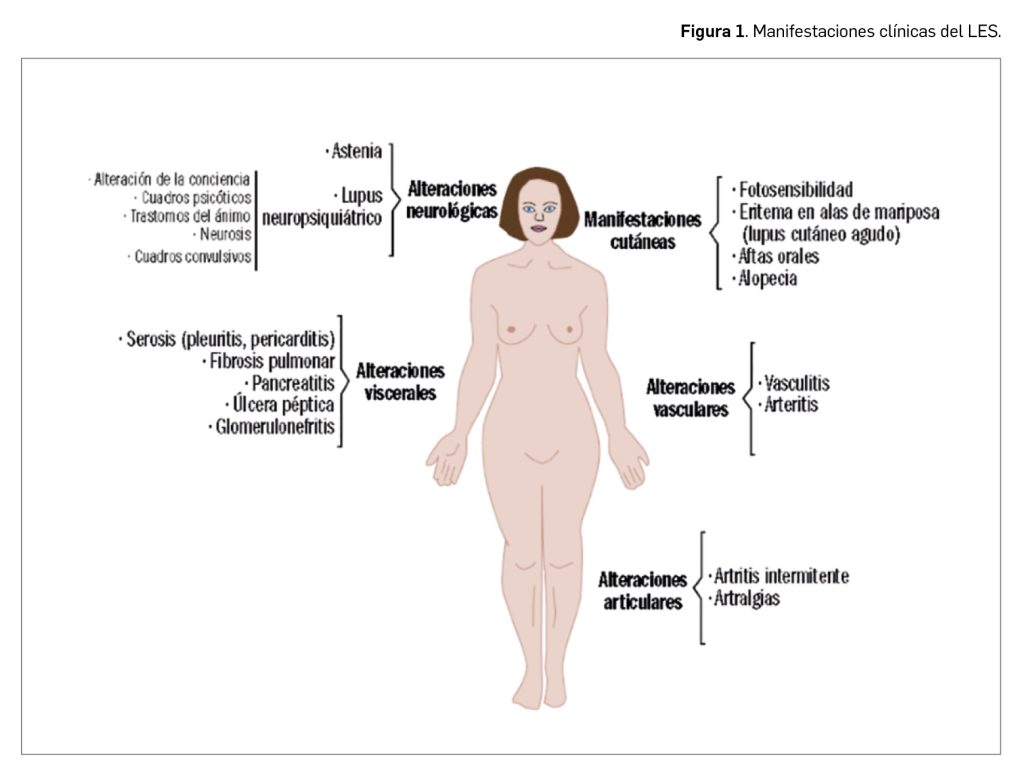
A nivel diagnóstico se recurre a una serie de criterios que permiten distinguir el LES de otras enfermedades autoinmunes en las que pueden presentarse manifestaciones clínicas similares, como la artritis reumatoide. Uno de los documentos más empleados es el elaborado de manera conjunta por la European Alliance of Associations for Rheumatology (EULAR) y el American College of Rheumatology (ACR) (Aringer et al., 2019), que puede consultarse en la Tabla 1. De acuerdo a estos criterios, se podrá considerar que un paciente padece LES cuando cumpla el criterio de entrada, al menos un criterio clínico y tenga una puntuación total igual o mayor a 10, considerando criterios clínicos e inmunológicos.

El LES afecta con mayor prevalencia a las mujeres, aunque con diferencias entre grupos de edad, lo que hace presumir que al menos hasta cierto grado existe en la etiopatogenia de la enfermedad un componente hormonal relacionado con los estrógenos. Además, la enfermedad suele tener una presentación bimodal: la mayoría de los casos se da en torno a los 30 años y otro pico menor en torno a los 55 años. Se estima que solo el 3-5% de los pacientes con LES cuenta con otros miembros familiares afectados.
Por otra parte, el pronóstico de la enfermedad ha cambiado sustancialmente: mientras que la supervivencia del LES a los 5 años del diagnóstico rondaba el 50% en 1955, actualmente ronda el 95% a los 5 años y el 93% a los 10 años. Los factores que más influyen en su evolución son la afectación aguda (peor cuanto más precoz) y crónica (daño acumulado) por la enfermedad. El mantenimiento de la inflamación crónica parece inducir la presencia acelerada de daño ateromatoso en las arterias, independientemente de la presencia de otros factores de riesgo vascular clásicos (hipertensión arterial, diabetes, tabaquismo, colesterol, etc.). También son importantes las complicaciones infecciosas, tanto por el uso de fármacos inmunosupresores como por la pérdida de las estructuras defensivas primarias por el efecto de la propia enfermedad (las alteraciones de la piel pueden favorecer las infecciones cutáneas, la presencia de hematuria y la proteinuria puede favorecer las infecciones urinarias, etc.). También la raza y la situación socioeconómica baja tienen repercusión en el pronóstico de estos pacientes: por ejemplo, la raza latina y la negra presentan peor supervivencia en LES.
Desde el punto de vista de su tratamiento, el objetivo terapéutico en pacientes con LES es disminuir el número y la duración de los brotes, evitando la pérdida y/o destrucción de tejidos con la menor toxicidad y efectos secundarios posibles. Además de evitar el daño agudo, es recomendable el control del proceso inflamatorio crónico, por los efectos deletéreos a largo plazo (afectación vascular, acumulación de daño orgánico, pérdida de calidad de vida, etc.).
En este sentido, hay dos grandes grupos de fármacos: los dirigidos a controlar la inflamación de una manera global a nivel tisular y aquellos que, de manera selectiva, inhiben un paso fundamental para el desarrollo del proceso inflamatorio, ya sea a nivel celular o humoral. Entre los primeros destacan los antiinflamatorios no esteroideos (AINE), los corticosteroides o los inmunosupresores clásicos (azatioprina, metotrexato, ciclofosfamida, etc.), mientras que en el segundo grupo se incluye principalmente a los fármacos biológicos, considerados fundamentales para el control de la enfermedad moderada o grave.
Estos últimos permiten la actuación sobre puntos específicos de las vías de señalización implicadas en la respuesta inmune, interfiriendo y deteniendo el proceso inflamatorio que perpetúa la enfermedad: en LES se ha visto que el principal elemento que contribuye a la etiopatogenia son los linfocitos B, de ahí el interés en producir fármacos que inhiban específicamente la actividad de este tipo celular, bien favoreciendo su lisis, como el rituximab, o bien inhibiendo los elementos que las activan, como es el caso de belimumab.
Rituximab, autorizado para su uso en artritis reumatoide, se está usando también en pacientes con LES o vasculitis graves (especialmente afectación renal), aunque tal indicación no está reconocida oficialmente, y en cualquier caso, en combinación con algún inmunosupresor (como el micofenolato). Por su parte, belimumab es un anticuerpo monoclonal humano recombinante de tipo IgG1λ que se une selectivamente e inhibe la actividad biológica del estimulador de linfocitos B soluble (BLyS), implicado en la promoción de la supervivencia y la diferenciación de los linfocitos B. Belimumab cuenta con indicación en el tratamiento adyuvante en pacientes adultos con LES activo, con autoanticuerpos positivos y con un alto grado de actividad de la enfermedad (por ejemplo, anti-ADNdc positivo y bajo nivel de complemento) a pesar del tratamiento estándar. Su acción inhibitoria sobre el BLyS en pacientes con LES permite reducir la cantidad de subpoblaciones seleccionadas de linfocitos B y de autoanticuerpos, y normalizar los niveles de inmunoglobulinas y del complemento.
Hay un tercer grupo de fármacos con potencial terapéutico, el de los inmunomoduladores, donde se incluirían sustancias capaces de influir sobre ciertos aspectos del sistema inmunitario, pero sin determinar inmunodepresión. El ejemplo más representativo de este grupo es la hidroxicloroquina1, un agente antipalúdico ampliamente utilizado en el tratamiento del LES, con acción lisosomotropa y capaz de inhibir el procesamiento antigénico.
Acción y mecanismo
Anifrolumab es un nuevo anticuerpo monoclonal que se une con alta afinidad y especificidad a la subunidad 1 del receptor del interferón de tipo 1 (IFNAR1). En condiciones fisiológicas, la unión del interferón de tipo 1 (IFN-1) a este receptor activa la cascada de señalización JAK-STAT, que regula la expresión de los genes relacionados con los interferones. La acción del fármaco permite reducir la producción de IFN-1, habitualmente sobreexpresado en pacientes con LES y que parece estar relacionado con la patogenia de la enfermedad debido a factores como la estimulación que esta molécula produce sobre la maduración de las células dendríticas, la producción de autoanticuerpos, la producción de inmunocomplejos o la inflamación orgánica.
Se comprende, por tanto, que el bloqueo de anifrolumab sobre los efectos del IFN-1 ha sido la base de la autorización de anifrolumab por la Agencia Europea de Medicamentos. Se encuentra comercializado en España con indicación en el tratamiento por vía intravenosa del lupus eritematoso sistémico (LES) con autoanticuerpos positivos, activo de moderado a grave en pacientes adultos, en combinación con el tratamiento estándar.
Aspectos moleculares
Anifrolumab es un anticuerpo monoclonal de tipo IgG1κ dirigido frente a la subunidad 1 del receptor del interferón de tipo 1 (IFNAR1). Cuenta con una masa molecular de aproximadamente 148 KDa, con dos cadenas pesadas idénticas entre sí de 49 KDa cada una y dos cadenas ligeras también idénticas entre sí de 23 KDa cada una. Se realizó una modificación sobre la región constante en tres aminoácidos con el objetivo de eliminar el receptor Fc gamma de tipo I/IIA/IIB/IIIA y la unión al componente C1q del complemento, lo cual permite eliminar la citotoxicidad celular dependiente de anticuerpos y la citotoxicidad dependiente del complemento.
Eficacia y seguridad clínicas
La eficacia y la seguridad clínicas de anifrolumab (300 mg por vía intravenosa cada 4 semanas) han sido examinadas en tres estudios clínicos, de los cuales se consideró pivotal el estudio TULIP-2, de fase 3, aleatorizado, doble ciego, multicéntrico y controlado con placebo, en el que participaron pacientes adultos con LES crónico de moderado a grave a pesar de recibir el tratamiento estándar. Los estudios TULIP-1, también de fase 3, y MUSE-2, de fase 2, se consideraron de soporte2.
En TULIP-2, los participantes (N= 362) recibieron 300 mg de anifrolumab por vía intravenosa o placebo (1:1) cada 4 semanas durante 48 semanas, con una duración total del estudio de 52 semanas. Tanto en este estudio como en los de soporte se incluyeron pacientes diagnosticados de acuerdo a criterios ACR y con un tratamiento concomitante y estabilizado con un corticoide y un inmunosupresor, pero no con un fármaco biológico (belimumab). Con exclusión de pacientes con formas renales y neuropsiquiátricas graves de LES, las características sociodemográficas basales de los participantes estuvieron equilibradas entre los distintos brazos de tratamiento en el estudio pivotal y también fueron comparables en los estudios de soporte. Hubo mayoría de mujeres (> 90%) y la edad media fue de 39-43 años.
La variable principal de eficacia en el estudio pivotal fue la tasa de pacientes con respuesta en la semana 52 de acuerdo a criterios BICLA, tras no observarse una mejora significativa al utilizar criterios SRI-4 en TULIP-13. Entre los objetivos secundarios se incluyeron la eficacia en pacientes con elevada expresión del gen del IFN-1 –tasa de respondedores según BICLA en la semana 52–, la reducción de la dosis de corticoides, la tasa anualizada de brotes o la eficacia sobre las lesiones cutáneas.
De acuerdo a los resultados divulgados (Morand et al., 2019), la tasa de respuesta a anifrolumab fue significativamente superior a placebo en la semana 52 (47,8% vs 31,5%; p= 0,0013). En el grupo de pacientes con alta expresión del gen de IFN (83,1% del total), el resultado fue numéricamente similar a favor de anifrolumab y también fue una diferencia estadísticamente significativa (48,0% vs. 30,7%; p= 0,0022). Además, se observó que el 51,5% de los pacientes tratados con anifrolumab vs. el 30,2% de los tratados con placebo consiguieron reducir la dosis de corticoide a ≤ 7,5 mg/día de prednisona (o equivalente) hasta la semana 52 (p= 0,0135). En cambio, no hubo diferencias notables respecto a la tasa anualizada de brotes, aunque numéricamente la tendencia fue menor en el grupo de anifrolumab (0,43 vs. 0,64). Respecto a la eficacia sobre las lesiones cutáneas, la proporción de pacientes con una reducción en la semana 12 de al menos el 50% en la escala CLASI4 fue superior en el brazo de anifrolumab (49,0% vs. 25,0%; p= 0,0392) entre quienes tenían una puntuación basal ≥ 10 puntos.
Los estudios de soporte, también de 52 semanas de duración, contaron con objetivos similares a los del pivotal. Sin embargo, en TULIP-1 la dosis autorizada de anifrolumab no fue superior a placebo en términos de la variable principal de eficacia –con criterios SRI-4–, al obtenerse una tasa de respuesta del 36,1% en el brazo del nuevo fármaco vs. del 40,2% en el brazo de placebo. En cambio, en línea con lo descrito en TULIP-2, la respuesta según criterios BICLA sí fue favorable a anifrolumab (47,1% vs. 30,2%; p= 0,001) en un análisis post-hoc no ajustado. En el otro estudio de soporte de fase 2, aleatorizado, doble ciego y controlado con placebo, la respuesta con anifrolumab fue superior a placebo tanto con criterios BICLA (53,5% vs. 25,7%) como con criterios SRI-4 (62,6% vs. 40,2%).
Se dispone asimismo de los resultados de un estudio de extensión aleatorizado, doble ciego y controlado por placebo, de 3 años de duración, en el que participaron 547 pacientes que habían completado el periodo de 52 semanas en los estudios de fase 3. Si bien su objetivo principal fue evaluar la seguridad del fármaco a largo plazo, se analizaron con carácter exploratorio algunas variables de eficacia: el tratamiento con anifrolumab permitió durante este periodo la reducción de la dosis de corticoides, de manera que solo el 9,9% de los pacientes vs. el 29,3% del grupo de placebo requería de una dosis > 7,5 mg/día de prednisona (o equivalente) al final del periodo de extensión (Kalunian et al., 2023). La tasa anualizada de brotes al final del estudio fue de 0,1 en el grupo de anifrolumab frente a 0,2 en el grupo de placebo.
En cuanto al perfil de seguridad, durante su desarrollo clínico un total de 1029 participantes estuvieron expuestos al nuevo fármaco, 837 de ellos pacientes de LES que lo recibieron por vía intravenosa. La duración media de exposición a la dosis aprobada (300 mg) en los estudios de fase 3 (n= 360) fue de 761 días (vs. 487 para placebo; n= 365), con una proporción elevada de pacientes que sufrió algún evento adverso (88,3% vs. 80,8%). Las infecciones fueron el tipo de evento adverso más frecuente (71,7% en el brazo de anifrolumab vs. 57,8% con placebo), destacando entre las reacciones de incidencia superior con anifrolumab las nasofaringitis (17,8% vs. 11,2%), infecciones del tracto respiratorio superior (16,9% vs. 9,9%), bronquitis (10,6% vs. 4,7%), herpes zóster (6,4% vs. 1,4%) y artralgia (5,6% vs. 2,2%). No obstante, en su mayoría fueron leves-moderadas, y la frecuencia de eventos graves en los estudios de fase 3 fue inferior en los pacientes que recibieron la dosis de 300 mg de anifrolumab frente a placebo (11,1% vs. 16,4%).
El perfil de eventos adversos a largo plazo en el estudio de extensión fue similar al de los estudios TULIP-1 y TULIP-2, con una tendencia temporal a la reducción de los eventos graves, desde el 11,8% de los pacientes en el año 1 hasta un 5,6% al final del periodo de extensión. En global, se reportaron 9 fallecimientos durante la evaluación clínica de anifrolumab, de los cuales 2 se consideraron debidos a eventos relacionados con el fármaco: en concreto, dos casos de neumonía, uno de los cuales se produjo en el estudio pivotal. Otros eventos adversos considerados de especial interés han sido las reacciones de hipersensibilidad, en su mayoría leves, de aparición durante las primeras 24 semanas de tratamiento y más comunes entre los pacientes tratados con anifrolumab (3,3% vs. 0,8%), y un posible aumento del riesgo de neoplasias5 que, por su relevancia y relación con el mecanismo de acción del fármaco, requiere de un seguimiento a largo plazo.
Aspectos innovadores
Anifrolumab es un nuevo anticuerpo monoclonal de tipo IgG1κ que se une con alta afinidad y especificidad a IFNAR1. En condiciones fisiológicas, la unión del interferón de tipo 1 (IFN-1) a este receptor activa la cascada de señalización JAK-STAT, que entre otras acciones regula la expresión de los genes relacionados con los interferones. Su acción bloqueante sobre los receptores permite al nuevo fármaco reducir la producción de IFN-1, habitualmente sobreexpresado en pacientes con LES y que parece estar relacionado con la patogenia de la enfermedad al estimular la maduración de las células dendríticas, la producción de autoanticuerpos, la producción de inmunocomplejos o la inflamación orgánica. En base a ello, anifrolumab ha sido autorizado para el tratamiento por vía intravenosa del LES con autoanticuerpos positivos, activo de moderado a grave en pacientes adultos, en combinación con el tratamiento estándar.
Su eficacia superior a placebo ha sido adecuadamente contrastada en el estudio pivotal TULIP-2, un fase 3, aleatorizado, doble ciego, multicéntrico y controlado con placebo, en el que participaron pacientes adultos con LES crónico de moderado a grave a pesar de recibir el tratamiento estándar. Los resultados revelaron que la tasa de pacientes con respuesta en la semana 52 de acuerdo a criterios BICLA (variable primaria) fue significativamente mayor con anifrolumab respecto a placebo (48% vs 32%; p= 0,0013), algo que se corroboró en igual medida en el subgrupo de pacientes con alta expresión del gen de IFN (83% del total). Las principales variables secundarias de eficacia arrojaron resultados también favorables al nuevo fármaco: por ejemplo, el tratamiento con anifrolumab permitió reducir la dosis de corticoides (a ≤ 7,5 mg/día de prednisona o equivalente) en mayor número de pacientes (52% vs. 30% con placebo) y se vio una mejora significativa de las lesiones cutáneas a la semana 12 en casi el doble de ellos (49% vs. 25% con placebo).
Dichos hallazgos se ven respaldados por los resultados a largo plazo de un estudio de fase 3 de extensión. Así, en pacientes que completaron los estudios de fase 3 de 1 año de duración se vio que la capacidad de anifrolumab para reducir la dosis de corticoides se mantuvo superior a placebo tras un total de 4 años de tratamiento: solo el 10% de los pacientes tratados requirió una dosis alta, frente al 29% en el grupo de placebo.
No obstante, los resultados de eficacia presentan ciertas limitaciones, entre las que destaca, en primer lugar, el planteamiento original del solicitante de incluir también el estudio TULIP-1 como pivotal, pero cuyo resultado negativo respecto a la variable primaria obligó a descartar tal opción y motivó el cambio desde el índice SRI-4 a BICLA como criterio para la variable principal. Por otro lado, en el estudio pivotal se registró una elevada tasa de discontinuación que podría afectar a la estimación del efecto, reduciendo la diferencia absoluta entre brazos desde el 16-17% hasta el 10-11% (EMA, 2022). Además, tampoco está claro si existen diferencias de relevancia clínica entre subgrupos de pacientes, pues la eficacia de anifrolumab parecia superior entre quienes tenían un perfil de niveles bajos en los componentes C3 o C4 del complemento o con anticuerpos anti-ADNdc positivos en la línea de base.
Finalmente, el perfil toxicológico del fármaco, bien caracterizado a investigar en profundidad el riesgo de carcinogenicidad a largo plazo, parece aceptable y clínicamente manejable. La mayoría de los pacientes tratados experimenta algún evento adverso, con una solo incidencia ligeramente superior a placebo (88% vs. 81%). Las reacciones adversas más frecuentes fueron las de tipo infeccioso, principalmente nasofaringitis (18% vs. 11%), infecciones del tracto respiratorio superior (17% vs. 10%), bronquitis (11% vs. 5%) y herpes zóster (6% vs. 1%), pero en su mayoría leves-moderadas, pues los eventos graves tuvieron mayor incidencia con placebo (16% vs. 11%). En cualquier caso, el carácter inmunosupresor asociado al mecanismo de acción de anifrolumab se añade a los efectos del tratamiento que habitualmente se utiliza de manera concomitante en pacientes con LES, como los corticosteroides o los inmunosupresores clásicos, lo que implicará un mayor riesgo de infecciones. Pese a que la vacunación de estos pacientes estaría especialmente recomendada, se desconoce aún el efecto que anifrolumab puede ejercer sobre el desarrollo de una adecuada respuesta inmunitaria tras la vacunación.
No se dispone por ahora de comparaciones directas ni indirectas que evalúen la eficacia de anifrolumab respecto a otros fármacos biológicos usados en LES, lo que dificulta su posicionamiento. En una revisión sistemática a partir de 14 estudios clínicos en los que se evaluó de forma separada la seguridad de belimumab, rituximab o anifrolumab, se encontró una mayor asociación de estos dos últimos con la incidencia de herpes zóster en comparación con belimumab, pero no un mayor riesgo de infecciones por el virus de la gripe (Steiger et al., 2022).
En resumen, anifrolumab incorpora un nuevo mecanismo de acción en el tratamiento del LES. Dada la escasez de tratamientos específicamente dirigidos a dianas implicadas en la patogenia de la enfermedad y teniendo en cuenta el alcance de la toxicidad asociada al tratamiento crónico con corticosteroides e inmunosupresores, se puede considerar que, aunque de magnitud escasa, la eficacia de anifrolumab resultaría de relevancia clínica en pacientes adultos con LES activo y grave. Aunque sus efectos a largo plazo no se conocen completamente y requieren de seguimiento por su carácter inmunosupresor, los datos disponibles parecen apuntar a un perfil de seguridad más benigno que el asociado a dosis altas de corticoides6. De acuerdo con los datos disponibles, el IPT de la AEMPS indica que anifrolumab se posicionaría como una alternativa a belimumab en pacientes adultos sin complicaciones renales o neuropsiquiátricas.