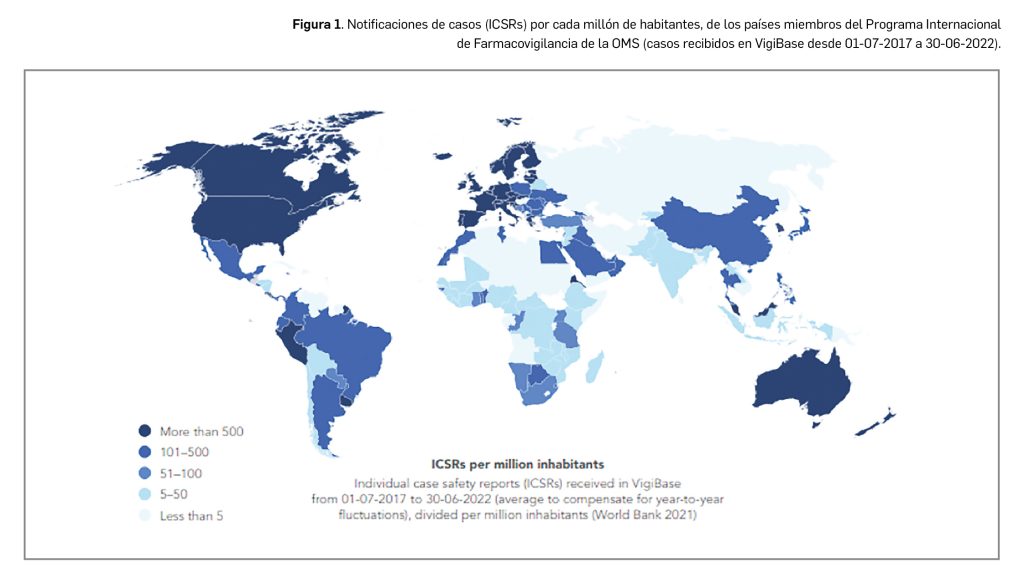
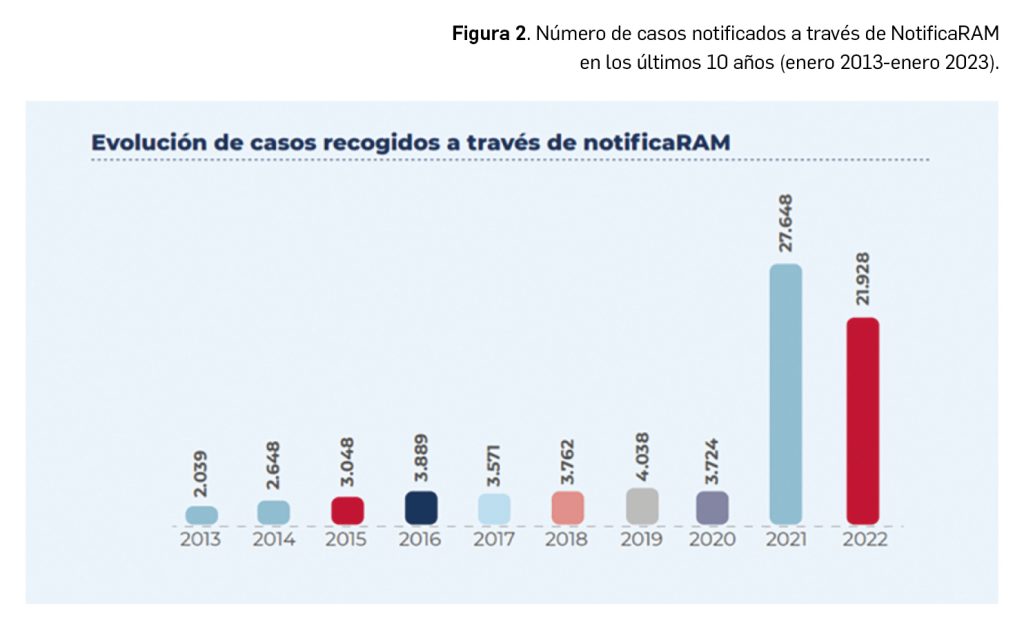
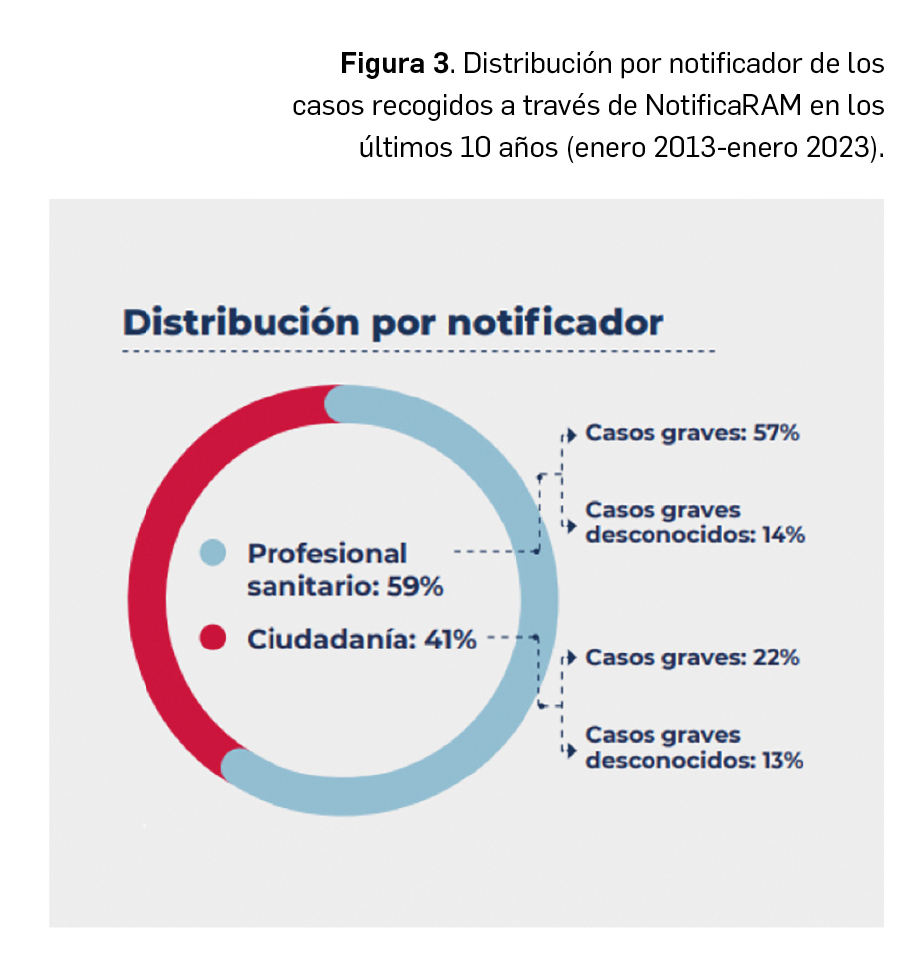
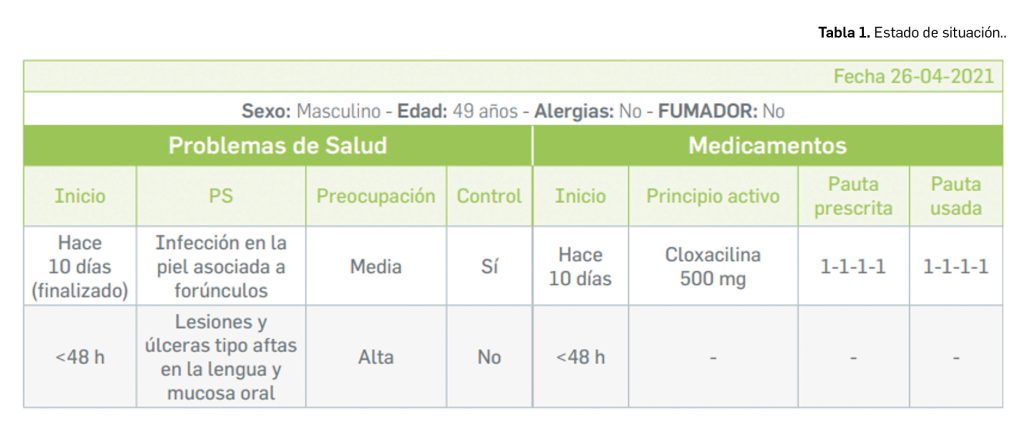
La agencia japonesa de medicamentos (PMDA) y su Ministerio de Salud, Trabajo y Bienestar (MHLW) han comunicado el riesgo de casos de anafilaxia asociados a la utilización de la vacuna frente al rotavirus humano, Rotarix®.
Se debe disponer del tratamiento médico apropiado en el caso de que ocurra un episodio anafiláctico tras la administración de la vacuna.
La agencia japonesa de medicamentos, Pharmaceuticals and Medical Devices Agency (PMDA) y su Ministerio de Salud, Trabajo y Bienestar (MHLW) han anunciado que la información del medicamento con la vacuna oral viva atenuada contra el rotavirus humano, Rotarix®, debe actualizarse para incluir el riesgo de anafilaxia (PMDA, 2023).
Esta vacuna oral viva atenuada contra el rotavirus humano está indicada para la prevención de la gastroenteritis causada por el rotavirus.
El ministerio (MHLW) y la agencia (PMDA) japoneses evaluaron 28 casos de notificaciones de eventos adversos nacionales relacionados con la vacuna y la anafilaxia, y en dos casos era razonablemente posible una relación causal entre la vacuna y el evento.
El MHLW y la PMDA concluyeron que la anafilaxia debe agregarse como una reacción adversa clínicamente significativa.
En España, la situación con la vacuna Rotarix® es la misma, pues no indica esta posible reacción de anafilaxia ni advertencia alguna. Por el contrario, la otra vacuna que existe frente al rotavirus, Rotateq®, con virus vivos de otras cepas, también producidas en células Vero, ya incluye información sobre esta posible reacción de anafilaxia. Y entre sus advertencias se describe que como con todas las vacunas, se debe disponer siempre del tratamiento médico adecuado en el caso de que ocurra un cuadro anafiláctico tras la administración de la vacuna (AEMPS, 2023).
Recomendaciones
Los profesionales sanitarios deben ser conscientes de este riesgo potencial y se les recomienda que, como con todas las vacunas, siempre se debe disponer del tratamiento médico apropiado en el caso de que ocurra un episodio anafiláctico tras la administración de la vacuna.