Resumen
Fedratinib es un nuevo inhibidor de la proteína JAK2 y de la tirosina cinasa 3 similar a FMS (FLT3), de tipo natural y mutada. FLT3 es una proteína con actividad tirosina cinasa relacionada con la diferenciación, proliferación y supervivencia de las células progenitoras hematopoyéticas. Las mutaciones que conducen a su actividad constitutiva promueven la proliferación celular y la resistencia a la apoptosis. Su actividad inhibidora sobre JAK2 permite reducir la fosforilación y la consiguiente activación de las proteínas activadoras de la transcripción STAT3/5. En base a estas acciones, fedratinib ha sido autorizado y comercializado en un medicamento designado como huérfano, con indicación en el tratamiento de la esplenomegalia o los síntomas relacionados con la enfermedad en pacientes adultos con mielofibrosis primaria, mielofibrosis posterior a policitemia vera o mielofibrosis posterior a trombocitemia esencial que no han recibido inhibidores de la cinasa asociada a Janus (JAK) previamente o han recibido tratamiento con ruxolitinib.
La eficacia de fedratinib fue evaluada en dos estudios pivotales. El primero de ellos (N= 289) fue un estudio de fase 3, multicéntrico, aleatorizado, doble ciego y controlado por placebo, en pacientes con mielofibrosis (MF) de riesgo intermedio 2 o alto y esplenomegalia. Su objetivo principal fue la evaluación de la tasa de respuesta (TR) al tratamiento con fedratinib (proporción de pacientes con una reducción del volumen del bazo (RVB) ≥ 35% tras recibir 6 ciclos de tratamiento). Los resultados indican una TR del 37% con fedratinib 400 mg vs. el 1% de los pacientes en el brazo de placebo, con una diferencia estadísticamente significativa (p < 0,0001).
Asimismo, se llevó a cabo un estudio pivotal de fase 2, no aleatorizado, abierto y de un solo brazo en el que se evaluó la eficacia de fedratinib en pacientes sintomáticos de riesgo intermedio 1 y en pacientes de riesgo intermedio 2 o alto con MF, con el objetivo principal de evaluar la proporción de pacientes con una RVB ≥ 35% al final del ciclo 6 de tratamiento en pacientes previamente tratados con ruxolitinib. En el 48% de los pacientes para los que se disponía de respuesta en cualquier momento del estudio se observó una RVB ≥ 35%. Sin embargo, debido al diseño de un solo brazo, se realizó una evaluación más conservadora considerando únicamente como respuesta aquellos casos en que el dato correspondía a la evaluación tras el ciclo 6 de tratamiento. En este caso, la TR fue del 36%.
Los eventos adversos más comunes con fedratinib 400 mg en los estudios pivotales fueron diarrea (68% vs. 16% con placebo), náuseas (62% vs. 16%), anemia (52% vs. 14%) y vómitos (45% vs. 5%). La diarrea (57%), las náuseas (56%), los vómitos (41%) y la anemia (33%) fueron los eventos relacionados con el tratamiento más frecuentes. En cuanto a los eventos adversos de grado 3 o 4, los más comunes fueron anemia (41% vs. 7% con placebo) y trombocitopenia (17% vs. 6%). Tanto la anemia (26%) como la trombocitopenia (12%) fueron asimismo los eventos de grado 3 o 4 relacionados con el tratamiento más frecuentemente observados.
Entre las limitaciones de los dos estudios pivotales destaca su corta duración, motivada por la aparición de casos de encefalopatía, así como la ausencia de comparador activo y la falta de datos respecto a la supervivencia.
Fedratinib no incorpora un nuevo mecanismo de acción, aunque la inhibición dual de JAK2 y FLT3 es una aproximación innovadora en el tratamiento de la MF. Su perfil de eficacia y seguridad parece comparable al de ruxolitinib, con algunas diferencias relacionadas con la mayor selectividad del nuevo fármaco por JAK2. A pesar de las incertidumbres respecto a la duración de la respuesta y al riesgo de aparición de encefalopatía, fedratinib se posiciona como la principal alternativa en el manejo de la esplenomegalia asociada a MF en pacientes refractarios o no candidatos a recibir ruxolitinib.
Aspectos fisiopatológicos
Las enfermedades mieloproliferativas crónicas comparten la característica de ser clonales, es decir, se originan en una célula madre de la médula ósea con capacidad de diferenciación pluripotente. En dichas patologías se produce una proliferación, diferenciación y maduración efectiva de las células en la médula ósea, generando una médula hipercelular y un número incrementado de células maduras en sangre periférica; asimismo, es frecuente encontrar hepatoesplenomegalia debida a hematopoyesis extramedular o al secuestro de los hematíes en el bazo hipertrofiado (Álvarez et al., 2017).
Según la clasificación de la Organización Mundial de la Salud, los síndromes mieloproliferativos crónicos incluyen a la leucemia mieloide crónica Filadelfia+ BCR/ABL+, la leucemia neutrofílica crónica, la leucemia eosinofílica crónica/síndrome hipereosinofílico, la policitemia vera, la trombocitemia esencial y la mielofibrosis crónica idiopática, junto con otras enfermedades mieloproliferativas inclasificables.
En concreto, la mielofibrosis primaria (MFP) se caracteriza por una intensa fibrosis de la médula ósea, esplenomegalia, hematopoyesis extramedular y leucoeritroblastosis (granulocitos y eritrocitos inmaduros) en sangre periférica y glóbulos rojos en forma de lágrima (dacriocitos). En su fase temprana, se caracteriza por un número elevado de células CD34+ en la médula, mientras que las fases posteriores de fibrosis medular muestran una disminución de células CD34+ en la médula y un aumento de congestión esplénica y hepática con células CD34+. Además, hay formas secundarias de mielofibrosis, que derivan secundariamente de otros trastornos mieloproliferativos, especialmente la policitemia vera y la trombocitemia esencial1.
La mielofibrosis (MF) se origina por una mutación somática en una célula madre hematopoyética pluripotente, lo que le confiere ciertas ventajas proliferativas respecto a los progenitores normales. Esa población celular anormal libera diversas citocinas y factores de crecimiento (PDGF, TGF-β, VEGF, bFGF y calmodulina) en la médula, dando lugar a fibrosis, osteoesclerosis y angiogénesis, y la colonización de órganos y sitios extramedulares, como el hígado y el bazo, en los que produce un sobrecrecimiento (hepato y esplenomegalia).
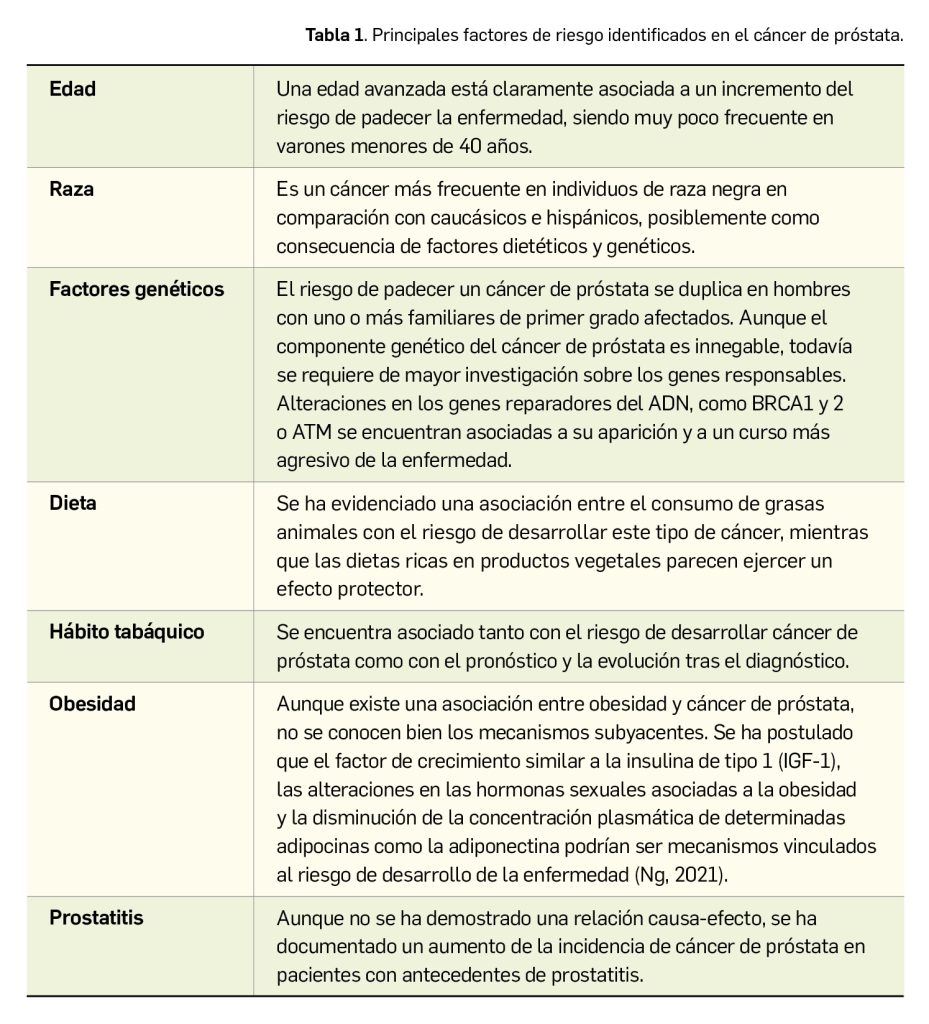
Presenta una incidencia de 4-14 casos por millón de habitantes/año, con una mediana de la edad de presentación de 65 años, aunque aproximadamente la tercera parte de los pacientes están asintomáticos al diagnóstico. Se suele diagnosticar entre los 50 y 80 años (menos del 10% son menores de 45 años), pero puede ocurrir a cualquier edad, y afecta tanto a hombres como a mujeres.
Aunque en muchos casos se ignora la etiología de esta enfermedad, en un 50% de los casos está presente una mutación del gen JAK2 y en un 10% una mutación del gen MPL2. La expresión de los genes JAK conduce a la síntesis de la familia de tirosina cinasas Janus (Janus tyrosine kinases, JAK), un miembro de la superfamilia de los receptores de citocinas. Las JAK parecen ejercer un papel esencial en los procesos de transducción de señales bioquímicas de diversas citocinas; de hecho, la eritropoyetina, la trombopoyetina y el factor de estimulante de colonias de granulocitos y macrófagos (GM-CSF) actúan exclusivamente sobre receptores que utilizan homodímeros de JAK2 en sus procesos de señalización intracelular.
En respuesta a la activación del receptor de citocinas, estas tirosina cinasas (que no forman parte propiamente de dicho receptor) son rápidamente fosforiladas dando lugar a una activación celular en cascada que supone la activación de varias vías de transducción de señales que determinan la aparición de cambios en la expresión génica que promueven la progresión y maduración celular.
La mutación de JAK2 no parece ser la causa primaria de la enfermedad sino más bien una mutación somática secundaria. Aunque la mutación JAK2 V617F3 parece ser la más común (aproximadamente, el 65%) de las asociadas a la MFP, se han identificado otras mutaciones que activan anormalmente al receptor JAK2, como la MPLW515L/Kin del receptor codificado por MPL. Sea como fuere, los altos niveles de citocinas encontrados en los pacientes con mielofibrosis, particularmente de interleucina 6 (IL-6), parecen ser los responsables del estado hipercatabólico y de los síntomas constitucionales de la enfermedad (pérdida de peso, fatiga, etc.). Muchas de estas citocinas utilizan tirosina cinasas de tipo JAK1 en su mecanismo señalizador celular.
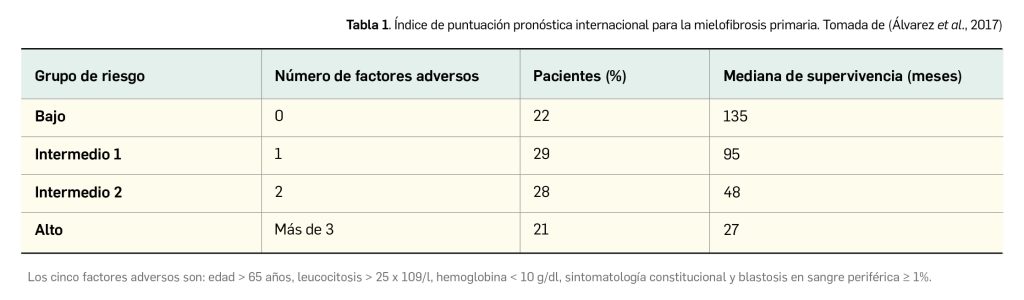
Las principales causas de muerte en los pacientes con mielofibrosis son la insuficiencia medular evolutiva, la transformación a leucemia no linfoblástica aguda, las infecciones oportunistas, las complicaciones trombo-hemorrágicas, la insuficiencia cardiaca y la hipertensión portal. Los factores pronósticos más relevantes son:
- Edad de 65 años o más.
- Anemia (hemoglobina < 10 g/dl). Síntomas inespecíficos: fiebre, sudores nocturnos o pérdida de peso. Leucocitosis (> 25 × 109/l).
- ≥ 1% de blastocitos circulantes.
La mediana de supervivencia en los pacientes con MFP es de 5-7 años (Alegre et al., 2017). Sin embargo, los pacientes sin ninguna característica adversa, excepto la edad, tienen una mediana de supervivencia de más de 10 a 15 años (mediana de 135 meses), pero la presencia de uno de los mencionados factores adversos la reduce a 95 meses (7,9 años), dos factores a 48 meses (4 años) y tres o más a 27 meses (2,25 años) (Tabla 1). Las anomalías cariotípicas también pueden afectar al pronóstico. En este sentido, algunas series retrospectivas han permitido observar que las deleciones 13q y 20q y la trisomía 9 se han correlacionado con una mayor supervivencia y ninguna transformación leucémica, en comparación con el peor pronóstico de la trisomía 8, un cariotipo complejo, -7/7q-, i(17q), inv(3), -5/5q-, 12p- o el reordenamiento 11q23.
Los pacientes asintomáticos de riesgo bajo solo requieren un seguimiento clínico, sin ninguna intervención terapéutica, que solo estaría justificada en caso de aparición de anemia sintomática, leucocitosis marcada, sudores nocturnos abundantes, pérdida de peso, fiebre o esplenomegalia sintomática.
El trasplante alogénico de progenitores hematopoyéticos es el único tratamiento curativo de la mielofibrosis, pero su aplicabilidad está limitada por la avanzada edad de la mayoría de los pacientes y por la existencia de diversas condiciones comórbidas.
La hidroxiurea (o hidroxicarbamida) se utiliza como tratamiento citorreductor y tiene indicación específica en esplenomegalia. Sin embargo, solo permite obtener respuestas parciales y de corta duración (AEMPS, 2023). La introducción de ruxolitinib, un inhibidor selectivo de las tirosina cinasas de tipo Janus JAK1 y JAK2, permitió obtener mejores respuestas en los síntomas asociados a la esplenomegalia, aunque su utilidad clínica en la mejora de la supervivencia no está clara (EMA, 2021). Sin embargo, no se dispone de alternativas farmacoterapéuticas para los pacientes que no responden o en los que se pierde la respuesta a ruxolitinib.
Acción y mecanismo
Fedratinib es un nuevo inhibidor de la proteína JAK2 y de la tirosina cinasa 3 similar a FMS (FLT3), de tipo natural y mutada. FLT3 es una proteína con actividad tirosina cinasa relacionada con la diferenciación, proliferación y supervivencia de las células progenitoras hematopoyéticas, mientras que las mutaciones que conducen a su actividad constitutiva promueven la proliferación celular y la resistencia a la apoptosis. Su actividad inhibidora sobre JAK2 permite reducir la fosforilación y la consiguiente activación de las proteínas activadoras de la transcripción STAT3/5. En base a estas acciones, fedratinib ha sido autorizado y comercializado en un medicamento designado como huérfano, con indicación en el tratamiento de la esplenomegalia o los síntomas relacionados con la enfermedad en pacientes adultos con mielofibrosis primaria, mielofibrosis posterior a policitemia vera o mielofibrosis posterior a trombocitemia esencial que no han recibido inhibidores de la cinasa asociada a Janus (JAK) previamente o han recibido tratamiento con ruxolitinib.
En estudios preclínicos se pudo comprobar la mayor selectividad de fedratinib sobre el receptor JAK2 respecto al resto de proteínas de la familia de las tirosina cinasas de Janus. No obstante, no se puede excluir la posibilidad de una cierta capacidad inhibitoria sobre JAK1 (IC50 (nM): 2,4 para JAK2 vs. 13 para JAK1).
Aspectos moleculares
Desde el punto de vista de su estructura química, fedratinib es el N-(1,-dimetiletil)-3-[(5-metil-2-[[4-[2-(1-pirrolidinil)etoxi]fenil]amino]-4-pirimidinil]amino] bencenosulfonamida, y se corresponde con la fórmula molecular C27H36N6O3S. Su masa molecular es de 524,7 g/mol.
La sustancia pura es un polvo sólido cristalino de color blanco o blanquecino, no higroscópico. Su solubilidad en agua es pH-dependiente, siendo libremente soluble en soluciones ácidas y prácticamente insoluble en solución tamponada neutra (pH 6,8).
Fedratinib es capaz de establecer interacciones por puente de hidrógeno con el residuo Leu932 y de tipo carbono-hidrógeno con los residuos Leu855 y Glu930 de JAK2, entre otro tipo de uniones débiles (Shawky et al., 2022).
Eficacia y seguridad clínicas
La eficacia y la seguridad clínicas de fedratinib han sido evaluadas en dos estudios pivotales. El primero de ellos (JAKARTA) se trató de un estudio de fase 3, multicéntrico, aleatorizado, doble ciego y controlado por placebo, con pacientes con mielofibrosis (MFP, MF post-policitemia vera o MF post-trombocitemia esencial) de riesgo intermedio 2 o alto y esplenomegalia.
El objetivo principal de este estudio fue la determinación del efecto de fedratinib en la reducción del volumen del bazo (RVB) al final de 6 ciclos de tratamiento4 en términos de la proporción de pacientes con una RVB ≥ 35% (tasa de respuesta, TR), confirmada en una exploración de seguimiento 4 semanas después. Entre los objetivos secundarios, se examinó la reducción de síntomas en base a la puntuación TSS calculada a partir del cuestionario MFSAF5 tras 6 ciclos y la duración de la respuesta.
En el estudio se aleatorizó a un total de 289 pacientes que no habían recibido tratamiento previo con un inhibidor de JAK en proporción 1:1:1 para recibir placebo, fedratinib 400 mg o fedratinib 500 mg en una única toma diaria por vía oral. 71 de los pacientes asignados aleatoriamente a placebo fueron realeatorizados para recibir fedratinib 400 mg o fedratinib 500 mg tras completar 6 ciclos o sufrir progresión de la enfermedad antes de este momento.
En cuanto a las características sociodemográficas de los pacientes, el 59% fueron hombres, mayoritariamente caucásicos (89%) y tenían en la línea de base una edad mediana de 65 años, con una mayor proporción de pacientes de 65 o menos años en el brazo de fedratinib 400 mg (64%) que en el brazo de placebo (46%) y de fedratinib 500 mg (51%).
En el caso de las características basales de la enfermedad, éstas estuvieron bien balanceadas entre los tres brazos. Sin embargo, una menor proporción de pacientes que recibieron la dosis de 400 mg del nuevo fármaco presentaba enfermedad de alto riesgo (41%) en comparación con aquellos que recibieron placebo o la dosis de 500 mg (52% en cada caso). El volumen mediano del bazo de los participantes en la línea de base fue de 2568 ml (normal: 215 ml).
De acuerdo a los resultados publicados (Pardanani et al., 2015; Pardanani et al., 2021), la tasa de respuesta (TR) en términos de RVB ≥ 35% confirmada cuatro semanas después de la finalización de 6 ciclos de tratamiento fue del 36,5% con fedratinib 400 (IC95%: 26,8 – 46,1%) y del 40,2% con la dosis de 500 mg (IC95%: 30,4 – 50,0%) vs. el 1,0% de los pacientes en el brazo de placebo (IC95%: 0 – 3,1%), siendo esta diferencia estadísticamente significativa en ambos casos en comparación con placebo (p < 0,0001). En ausencia de la confirmación a las 4 semanas, la TR fue sensiblemente superior tanto con la dosis de 400 mg (46,9%) como con la dosis más alta (49,5%), sin modificaciones en el brazo de placebo.
Para los pacientes realeatorizados a los brazos de fedratinib, la RVB fue del 28,6% con la dosis inferior y del 44,4% con la dosis de 500 mg tras 6 ciclos de tratamiento.
En relación al objetivo secundario de mejora de los síntomas, en los dos brazos de tratamiento activo se observó una respuesta clínica estadísticamente significativa en comparación con placebo. La proporción de pacientes con una reducción en la TSS ≥ 50% tras 6 ciclos fue del 8,2% en el brazo de placebo, del 39,6% con fedratinib 400 y del 34,1% con fedratinib 500 mg (p ≤ 0,001).
La duración mediana de la respuesta fue de 18,2 meses con la dosis inferior y de 19,7 meses con la dosis de 500 mg.
Se dispone asimismo de los resultados de otro estudio pivotal (JAKARTA2) de fase 2, no aleatorizado, abierto, de un solo brazo y multicéntrico en el que se evaluó la eficacia de fedratinib en pacientes sintomáticos de riesgo intermedio 1 y en pacientes de riesgo intermedio 2 o alto con MF (MFP, post-policitemia vera o post-trombocitemia esencial), cuyo objetivo fue evaluar la eficacia y la seguridad de la administración de fedratinib 400 mg en pacientes previamente tratados con ruxolitinib, en términos de la TR, definida como la proporción de pacientes con una RVB ≥ 35% al final del ciclo 6 de tratamiento. Aunque en el estudio participaron 97 pacientes, se dispuso de evaluación del volumen esplénico tanto en la línea de base como tras 6 ciclos en 83. Así, de acuerdo a los resultados publicados (Harrison et al., 2017) de estos 83 pacientes, en el 48,2% se observó una RVB ≥ 35% al final de 6 ciclos de tratamiento (IC95%: 37,1 – 59,4%), incluyendo en esta TR a todos los pacientes en los que la última observación disponible cumplía el requisito de RVB de al menos el 35% (análisis LOCF)6.
Debido al diseño de un solo brazo, se realizó una evaluación más conservadora considerando como “sin respuesta” a todos aquellos pacientes para los cuales no se dispusiera del dato de volumen esplénico tras 6 ciclos de tratamiento. En este caso, la TR fue del 36,1% (IC95%: 25,9 – 47,4%).
En cuanto al análisis de la seguridad de fedratinib, se dispone fundamentalmente de los datos de los pacientes expuestos a fedratinib en los dos estudios pivotales (N= 290). Por lo que respecta a la dosis aprobada de 400 mg, en los estudios pivotales estuvieron expuestos 203 pacientes durante una mediana de 35,6 semanas. El 63% estuvieron expuestos al menos 6 meses y el 38% al menos 12 meses.
Todos los pacientes que recibieron fedratinib y el 93,7% de los que recibieron placebo experimentaron al menos un evento adverso7. El 90,6% de los eventos observados se consideraron relacionados con el tratamiento (38,9% en aquellos que recibieron placebo). Los eventos adversos de grado 3 o 4 fueron frecuentes en el tratamiento con fedratinib 400 mg (68,0%; 49,8% relacionados con el tratamiento) y fueron superiores con la dosis no aprobada de 500 mg (78,4%; 66,0% relacionados con el tratamiento), frente al 30,5% (9,5% relacionados con el tratamiento) entre quienes recibieron placebo.
Los eventos adversos más comunes (frecuencia > 20%) con fedratinib 400 mg en los estudios pivotales fueron diarrea (67,5% vs. 15,8% con placebo), náuseas (61,6% vs. 15,8%), anemia (51,7% vs. 13,7%), vómitos (44,8% vs. 5,3%), trombocitopenia (21,7% vs. 8,4%) y fatiga (20,7% vs. 9,5%). De los pacientes que recibieron fedratinib 400 mg, la diarrea (56,7%), las náuseas (56,2%), los vómitos (40,5%) y la anemia (33,0%) fueron los eventos relacionados con el tratamiento más frecuentes.
En cuanto a los eventos adversos de grado 3 o 4, los más comunes fueron los relacionados con el sistema circulatorio y linfático, principalmente anemia (41,4% vs. 7,4% con placebo) y trombocitopenia (16,7% vs. 6,3%). Tanto la anemia (26,1%) como la trombocitopenia (11,8%) fueron asimismo los eventos de grado 3 o 4 relacionados con el tratamiento más frecuentemente observados.
El 5,9% de los pacientes tratados con fedratinib 400 mg en los estudios pivotales falleció durante el periodo de estudio (6,3% entre aquellos que recibieron placebo). Se registraron dos eventos adversos relacionados con el tratamiento como causa del fallecimiento en dos pacientes (shock cardiogénico y leucemia aguda; uno en cada paciente). El 24,1% de los pacientes tratados con esta dosis discontinuó el tratamiento a consecuencia de un evento adverso (vs. 8,4% con placebo), siendo el más común la trombocitopenia (3,0% vs. 0% con placebo).
Entre los eventos adversos de especial interés se consideró la encefalopatía grave (incluyendo encefalopatía de Wernicke), observada en 8 pacientes que recibían fedratinib (7 de ellos en una dosis de 500 mg), de los cuales uno falleció.
Aspectos innovadores
Fedratinib es un nuevo inhibidor de la proteína JAK2 y de la tirosina cinasa 3 similar a FMS (FLT3), de tipo natural y mutada. FLT3 es una proteína con actividad tirosina cinasa relacionada con la diferenciación, proliferación y supervivencia de las células progenitoras hematopoyéticas. Las mutaciones que conducen a su actividad constitutiva promueven la proliferación celular y la resistencia a la apoptosis. Su actividad inhibidora sobre JAK2 permite reducir la fosforilación y la consiguiente activación de las proteínas activadoras de la transcripción STAT3/5. En base a estas acciones, fedratinib ha sido autorizado y comercializado en un medicamento designado como huérfano, con indicación en el tratamiento de la esplenomegalia o los síntomas relacionados con la enfermedad en pacientes adultos con mielofibrosis primaria, mielofibrosis posterior a policitemia vera o mielofibrosis posterior a trombocitemia esencial que no han recibido inhibidores de la cinasa asociada a Janus (JAK) previamente o han recibido tratamiento con ruxolitinib.
La eficacia y la seguridad clínicas de fedratinib han sido evaluadas en dos estudios pivotales.
JAKARTA fue un estudio de fase 3, multicéntrico, aleatorizado, doble ciego y controlado por placebo, en el que participaron pacientes con mielofibrosis (MFP, MF post-policitemia vera o MF post-trombocitemia esencial) de riesgo intermedio 2 o alto y esplenomegalia. Su objetivo principal fue la evaluación de la TR al tratamiento con fedratinib en términos de la proporción de pacientes con una RVB ≥ 35% tras recibir 6 ciclos de tratamiento.
Tras aleatorizar a 289 pacientes en tres brazos de tratamiento (en proporción 1:1:1), se obtuvo una TR –en términos de RVB ≥ 35% confirmada cuatro semanas después de la finalización de 6 ciclos– del 37% con fedratinib 400 (IC95%: 27 – 46%) y del 40% con la dosis de 500 mg (IC95%: 30 – 50%) vs. el 1% de los pacientes en el brazo de placebo (IC95%: 0 – 3%), con una diferencia estadísticamente significativa en comparación con placebo en los dos casos (p < 0,0001).
En relación al objetivo secundario de la mejora de los síntomas, la proporción de pacientes con una reducción en la TSS ≥ 50% tras 6 ciclos fue del 8% en el brazo de placebo, del 40% con fedratinib 400 y del 34% con fedratinib 500 mg.
Por otro lado, JAKARTA2 fue un estudio de fase 2, no aleatorizado, abierto, de un solo brazo y multicéntrico en el que se evaluó la eficacia de fedratinib en pacientes sintomáticos de riesgo intermedio 1 y en pacientes de riesgo intermedio 2 o alto con MF (MFP, post-policitemia vera o post-trombocitemia esencial), llevado a cabo con el objetivo principal de evaluar la eficacia de la administración de fedratinib 400 mg en pacientes previamente tratados con ruxolitinib, en términos de la TR, definida como la proporción de pacientes con una RVB ≥ 35% al final del ciclo 6 de tratamiento.
En el 48% de los pacientes para los que se disponía de respuesta en cualquier momento del estudio se observó una RVB ≥ 35%. Sin embargo, debido al diseño de un solo brazo, se realizó una evaluación más conservadora considerando como “sin respuesta” a todos aquellos pacientes para los cuales no se dispusiera de un dato final de volumen esplénico tras 6 ciclos de tratamiento. En este caso, la TR fue del 36% (IC95%: 26 – 47%).
En los estudios pivotales, el tratamiento con fedratinib se asoció con una aparición generalizada de eventos adversos, aunque estos también fueron muy frecuentes con placebo (100% vs. 94%), siendo también elevada la frecuencia de eventos relacionados con el tratamiento (91% vs. 39% con placebo). Los eventos adversos más comunes (frecuencia > 20%) con fedratinib 400 mg en los estudios pivotales fueron diarrea (68% vs. 16% con placebo), náuseas (62% vs. 16%), anemia (52% vs. 14%), vómitos (45% vs. 5%), trombocitopenia (22% vs. 8%) y fatiga (21% vs. 10%). De los pacientes que recibieron fedratinib 400 mg, la diarrea (57%), las náuseas (56%), los vómitos (41%) y la anemia (33%) fueron los eventos relacionados con el tratamiento más frecuentes.
En cuanto a los eventos adversos de grado 3 o 4, los más comunes fueron anemia (41% vs. 7% con placebo) y trombocitopenia (17% vs. 6%). Tanto la anemia (26%) como la trombocitopenia (12%) fueron asimismo los eventos de grado 3 o 4 relacionados con el tratamiento más frecuentemente observados.
La aparición de casos de encefalopatía y encefalopatía de Wernicke es un aspecto preocupante de la seguridad de fedratinib. No obstante, el nuevo principio activo no produjo otros signos relevantes de toxicidad neurológica y es probable que los casos observados, 8 en total, se encuentren relacionados con un déficit de tiamina, por lo que podrían prevenirse con una adecuada monitorización y, en su caso, suplementación.
A este respecto, los casos de encefalopatía motivaron la finalización temprana de los dos estudios pivotales, lo que limita la interpretación sobre la duración de la respuesta y el conocimiento respecto a los datos de seguridad.
Otra limitación importante de los estudios pivotales es la ausencia de comparador activo, que impide establecer una comparación precisa con la principal alternativa disponible, ruxolitinib. Además, la ausencia de datos maduros respecto a la supervivencia global y a la supervivencia libre de progresión debido a la corta duración de los estudios es un factor especialmente importante en el contexto del tratamiento de una neoplasia maligna.
No obstante, a pesar de estas limitaciones, los efectos positivos observados con fedratinib, tanto en cuanto respecta a la esplenomegalia como a los síntomas asociados, son relevantes en el manejo de los pacientes con MF. La tasa de respuesta del 37% en la variable principal de eficacia es similar a la obtenida por ruxolitinib (~42%) en el estudio pivotal COMFORT-I (EMA, 2020), aunque se requiere de comparaciones directas o de datos a más largo plazo provenientes de la experiencia clínica con ambos fármacos para caracterizar su eficacia de forma comparada.
Fedratinib, como inhibidor de la cinasa asociada a Janus, no incorpora un mecanismo de acción totalmente novedoso, aunque la inhibición dual de JAK2 y FLT3 es una aproximación innovadora en el tratamiento de la MF. Su perfil de eficacia y seguridad parece comparable al de ruxolitinib, con algunas diferencias relacionadas con la mayor selectividad del nuevo fármaco por JAK2, como una mayor toxicidad gastrointestinal y menor toxicidad hematológica (EMA, 2020). A pesar de las incertidumbres respecto a la duración de la respuesta y al riesgo de aparición de encefalopatía, fedratinib se posiciona como la principal alternativa en el manejo de la esplenomegalia asociada a MF en pacientes refractarios o no candidatos a recibir ruxolitinib.
Valoración