Resumen
El registro centralizado europeo es un procedimiento ágil que permite una comercialización razonablemente rápida y científicamente rigurosa de medicamentos innovadores, lo que debería facilitar su acceso casi inmediato para todos los ciudadanos de los países integrantes de la Unión Europea. Sin embargo, la disponibilidad real de tales medicamentos para el usuario –paciente– final no es tan inmediata, debido principalmente a que, tras la autorización europea de comercialización, cada estado miembro de la UE debe proceder a registrar el medicamento, estableciendo sus condiciones propias, entre otras las relativas al ámbito de dispensación y, muy particularmente, a su precio de venta y, eventualmente, su financiación por el sistema sanitario público. En concreto, el tiempo para la disponibilidad real de terapias innovadoras en España entre 2017 y 2020 era de una media de 517 días. En el presente artículo se ha procedido a revisar sistemáticamente y actualizar los datos correspondientes a 2021 y 2022, comprobando que de los 114 medicamentos aprobados por la Comisión Europea durante ese periodo, solo se han comercializado en España 32 (28%), lo que supone que aún en el caso de que los restantes fuesen comercializados el 4 de abril de 2023 (fecha del análisis) o posteriormente, lo serían con un retraso de, al menos, entre 105 y 817 días, con un promedio de 402 y una mediana de 361. Por su parte, el retraso en la comercialización en España de los medicamentos huérfanos aprobados por la Comisión Europea es particularmente significativo, ya que de los 41 medicamentos aprobados durante 2021 y 2022, solo se han comercializado en España 4 (10%), lo que en el caso de que los restantes fuesen comercializados el 4 de abril de 2023 o posteriormente, lo serían con un retraso de, al menos, entre 109 y 817 días, con un promedio de 394 y una mediana de 365. En definitiva, el retraso en el acceso en España a los medicamentos novedosos –y, en particular, los medicamentos huérfanos– autorizados por la Comisión Europea sigue siendo ciertamente preocupante en la actualidad (2023).
Introducción
El procedimiento centralizado permite comercializar un medicamento basándose en una única evaluación europea, mediante una autorización que es válida en toda la Unión Europea (en adelante, UE). El uso de este procedimiento centralizado es legalmente obligatorio para todos los países de la UE para la autorización de prácticamente cualquier medicamento innovador –incluyendo a los indicados para enfermedades raras (medicamentos huérfanos)– aunque la empresa titular no quiera comercializarlos en todos y cada uno de los países.
Con este procedimiento, las empresas farmacéuticas solo requieren presentar una única solicitud de autorización a la Agencia Europea de Medicamentos (Euroepan Medicines Agency; EMA). Tras ello, el Comité de Medicamentos de Uso Humano (Committee for Medicinal Products for Human Use; CHMP) o, en su caso, el Comité de Medicamentos de Uso Veterinario (Committee for Veterinary Medicinal Products; CVMP) de la EMA revisa y evalúa la documentación administrativa y científica que acompaña a la solicitud, tras lo que formula una recomendación a la Comisión Europea (European Comission, EC) sobre si se puede conceder o no una autorización de comercialización para todos los Estados miembros de la UE. La decisión que toma la citada Comisión, en base a la recomendación de la EMA, es la que tiene carácter legal vinculante.
Un acceso a los medicamentos innovadores… no tan rápido
En principio, el registro centralizado europeo es un procedimiento ágil que permite una comercialización razonablemente rápida y científicamente rigurosa de medicamentos innovadores útiles para el tratamiento, prevención y diagnóstico de enfermedades y síndromes, lo que debería facilitar su acceso casi inmediato para todos los ciudadanos de los países integrantes de la UE.
Sin embargo, la disponibilidad real de los medicamentos para el usuario final no es tan inmediata como parece sugerir el procedimiento centralizado. El principal motivo de ello es que, tras la autorización europea de comercialización, cada país de la UE debe proceder a registrar el medicamento –se ahorran, eso sí, todo el complejo proceso de evaluación, ya realizado por la EMA– estableciendo sus condiciones propias, entre otras las relativas al ámbito de dispensación y, muy particularmente, a su precio de venta y, eventualmente, su financiación –total, parcial o nula– por el sistema sanitario público.
Según los datos del Informe Wait, recogido por la European Federation of Pharmoaceutical Industries and Associations (EFPIA), en el caso de España el tiempo para la disponibilidad real de terapias innovadoras era de una media de 517 días, durante el periodo comprendido entre 2017 y 2020. Esto sitúa a España en las posiciones intermedias de los países europeos, ya que mientras que en Alemania y Dinamarca este periodo era de 133 y 176 días, respectivamente, en Rumanía asciende a los 899.
Según el citado informe Wait, es difícil cuantificar el impacto total de los retrasos, pero es razonable pensar que pueden conducir a una mayor mortalidad y, por tanto, son muertes evitables; pero también inciden en la pérdida de calidad de vida para los pacientes y para sus familiares y cuidadores; suponen un impacto real en los costes de la atención sanitaria, que podría haber sido evitado con tratamientos más novedosos (supuestamente más efectivos clínicamente y eficientes económicamente), e impactan en cadena sobre los recursos disponibles para otros pacientes. Por otra parte, los retrasos en la disponibilidad de medicamentos novedosos también inducen indirectamente una pérdida de empleo productivo y, en última instancia, un mayor esfuerzo para la economía nacional.
Por su parte, las autoridades sanitarias españolas aseguran que algo más de la mitad del tiempo transcurrido no es achacable a su gestión. De hecho, el Ministerio de Sanidad afirma que el tiempo medio desde la autorización de la Comisión Europea hasta que el laboratorio solicita la comercialización en nuestro país, y se le asigna un código nacional, registrándose así en la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS), es de 180,3 días, mientras que el tiempo medio desde que se inicia el estudio de financiación y precio hasta que el laboratorio titular presenta el dossier con la primera oferta de precio es de 107,3 días.
¿Y cuál es la situación actual?
Más allá de las estadísticas oficiales y empresariales, que suelen manejar valores promedios que muchas veces son poco o nada útiles para valorar las necesidades reales de cada paciente en concreto, el acceso en España a los medicamentos novedosos autorizados por la Comisión Europea es ciertamente preocupante.
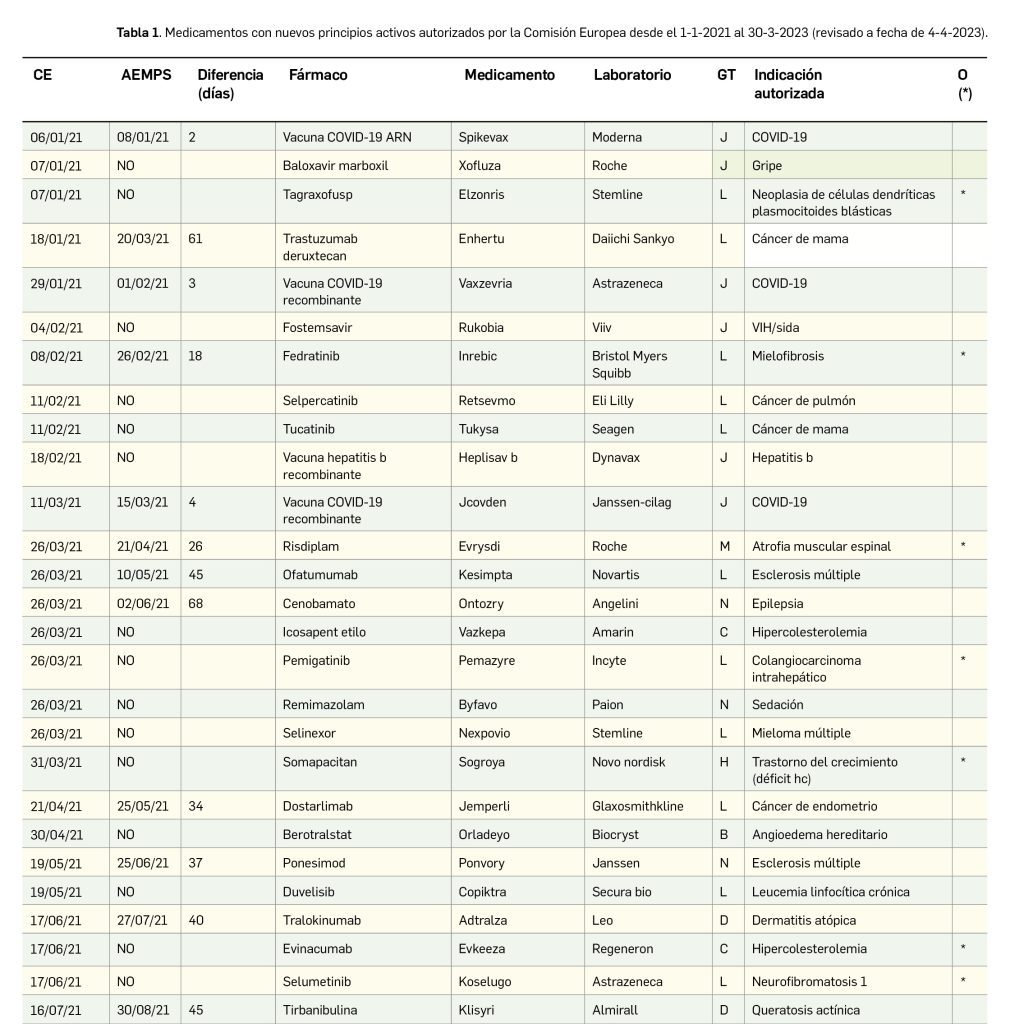
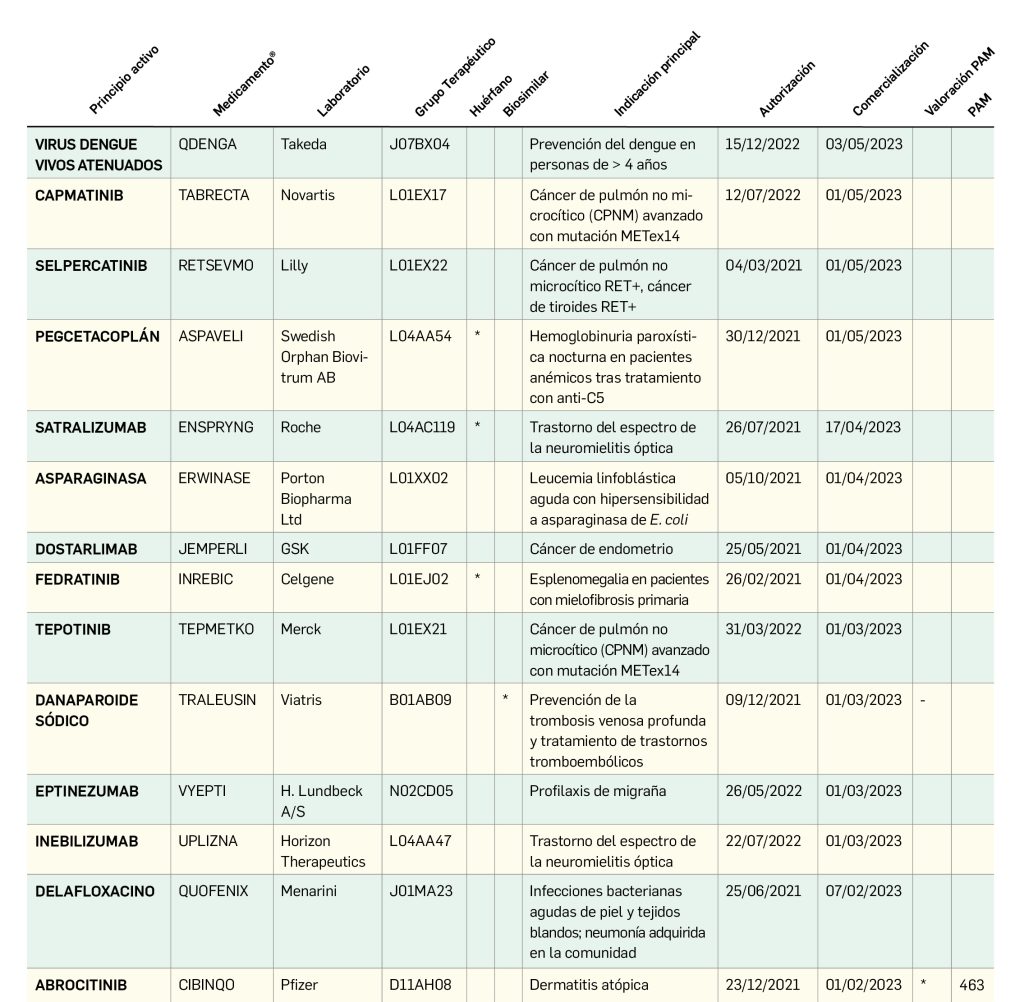
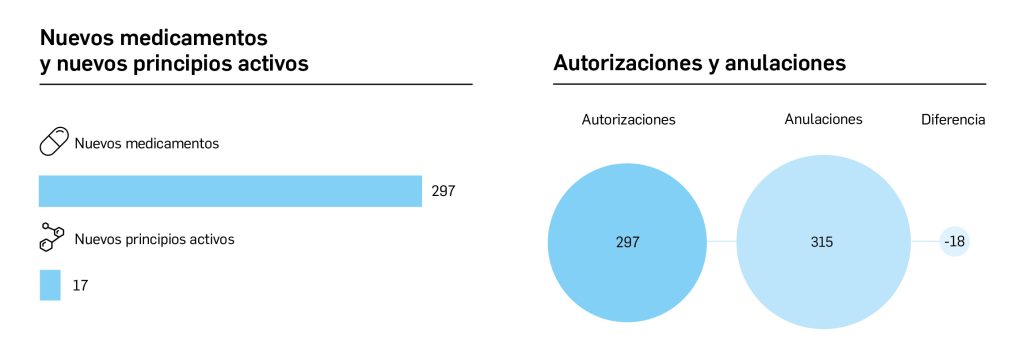
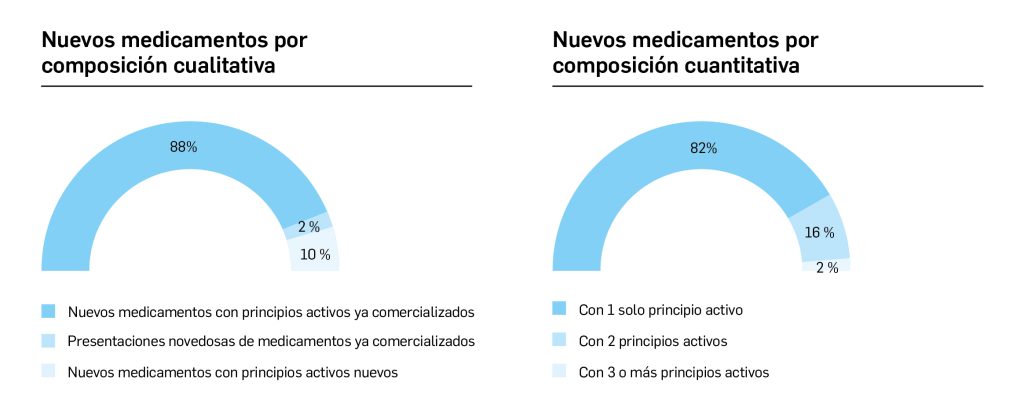
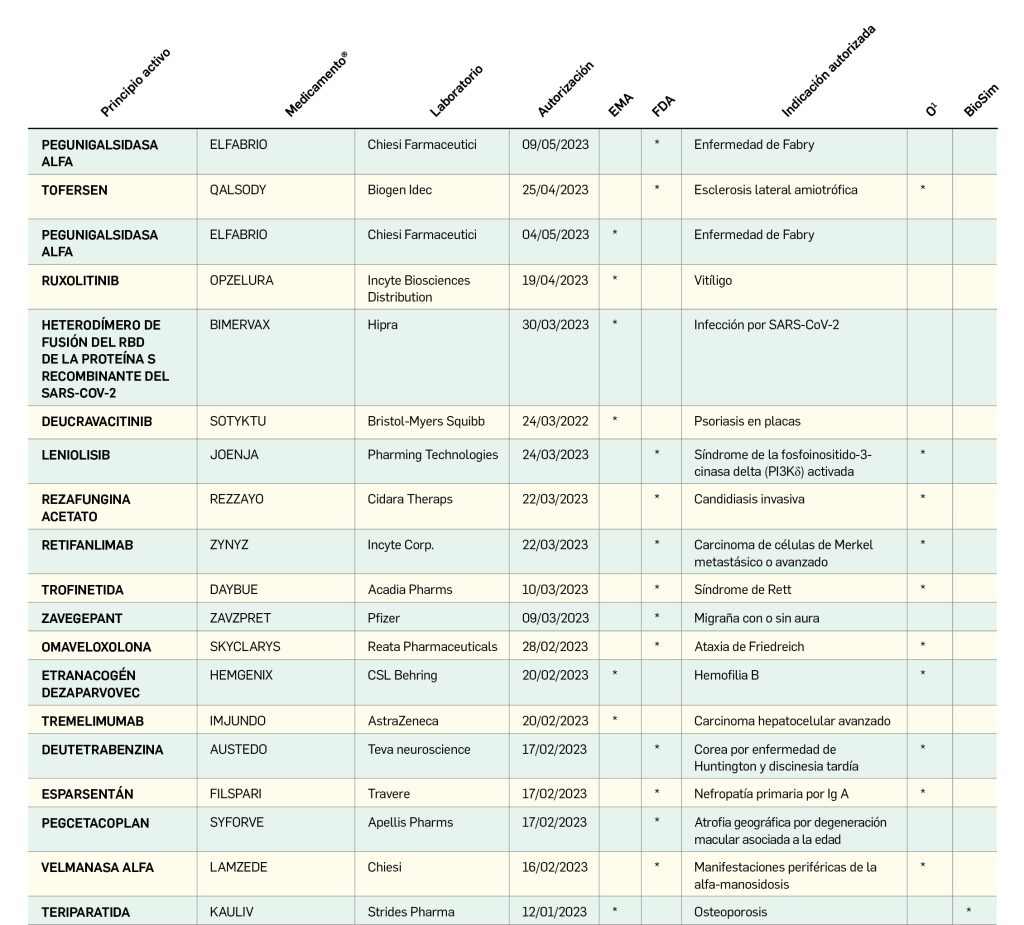
En el presente informe, cuyos datos están recogidos sistemáticamente en la Tabla 1 y preparados a partir de los procedentes del Centro de información online de medicamentos (CIMA) de la AEMPS y del Union Register of Medicinal Products de la Comisión Europea, actualizados a fecha de 4 de abril de 2023, se muestra que entre el 1 de enero de 2021 y el 30 de marzo de 2023, se autorizaron 119 medicamentos con nuevos principios activos, es decir, sin incluir biosimilares, ni genéricos, ni antiguos medicamentos con nuevas indicaciones autorizadas; de ellos, 41 (34% del total) son medicamentos huérfanos.
De los 119 medicamentos novedosos autorizados por la CE en ese periodo, solo 32 (27%) lo han sido por la AEMPS y comercializados en España, a fecha de 4 de abril de 2023, de los que solo 4 (13%) son huérfanos. Por otro lado, hay una diferencia entre la fecha de autorización de la Comisión Europea y la de la AEMPS de entre 1 y 178 días, con un promedio de 42 días y una mediana de 36; aunque, ciertamente, las vacunas y tratamientos anti-COVID solo tuvieron un retraso de 1 a 6 días. De los 41 medicamentos huérfanos autorizados por EMA en el periodo analizado, solo 4 (10%) lo han sido en España.
Previsiblemente, una parte sustancial de los medicamentos novedosos (medicamentos con nuevos principios activos) autorizados por la Comisión Europea desde el 1 de enero de 2021 y que aún no están comercializados en España, acabarán por estarlo aunque con un notable retraso. Así se evidencia a continuación, prescindiendo de los 5 autorizados por la CE durante el trimestre enero-marzo de 2023, para no sesgar los resultados del análisis estadístico:
- Globalmente, de los 114 medicamentos aprobados por la CE durante 2021 y 2022 (del 1 de enero de 2021 al 31 de diciembre de 2022), solo se han comercializado en España 32 (28%), al día 4 de abril de 2023 (fecha del análisis). Esto supone que aún en el caso de que los restantes fuesen comercializados el 4 de abril de 2023 o posteriormente, lo serían con un retraso de, al menos, entre 105 y 817 días, con un promedio de 402 y una mediana de 361.
- De los 50 medicamentos aprobados por la CE durante 2021, solo están comercializados actualmente en España 21 (42%). En este caso, el retraso sería de, al menos, entre 147 y 817 días, con un promedio de 615 y una mediana de 627.
- De los 64 medicamentos aprobados por la CE durante 2022, solo están comercializados en España 12 (19%). En este caso, el retraso sería de, al menos, entre 105 y 448 días, con un promedio de 269 y una mediana de 260.
El retraso en la comercialización en España de los medicamentos huérfanos aprobados por la Comisión Europea es particularmente significativo, ya que de los 41 medicamentos aprobados por la CE durante 2021 y 2022, solo se han comercializado en España 4 (10%). Esto supone que aún en el caso de que los restantes fuesen comercializados el 4 de abril de 2023 o posteriormente, lo serían con un retraso de, al menos, entre 109 y 817 días, con un promedio de 394 y una mediana de 365 días.
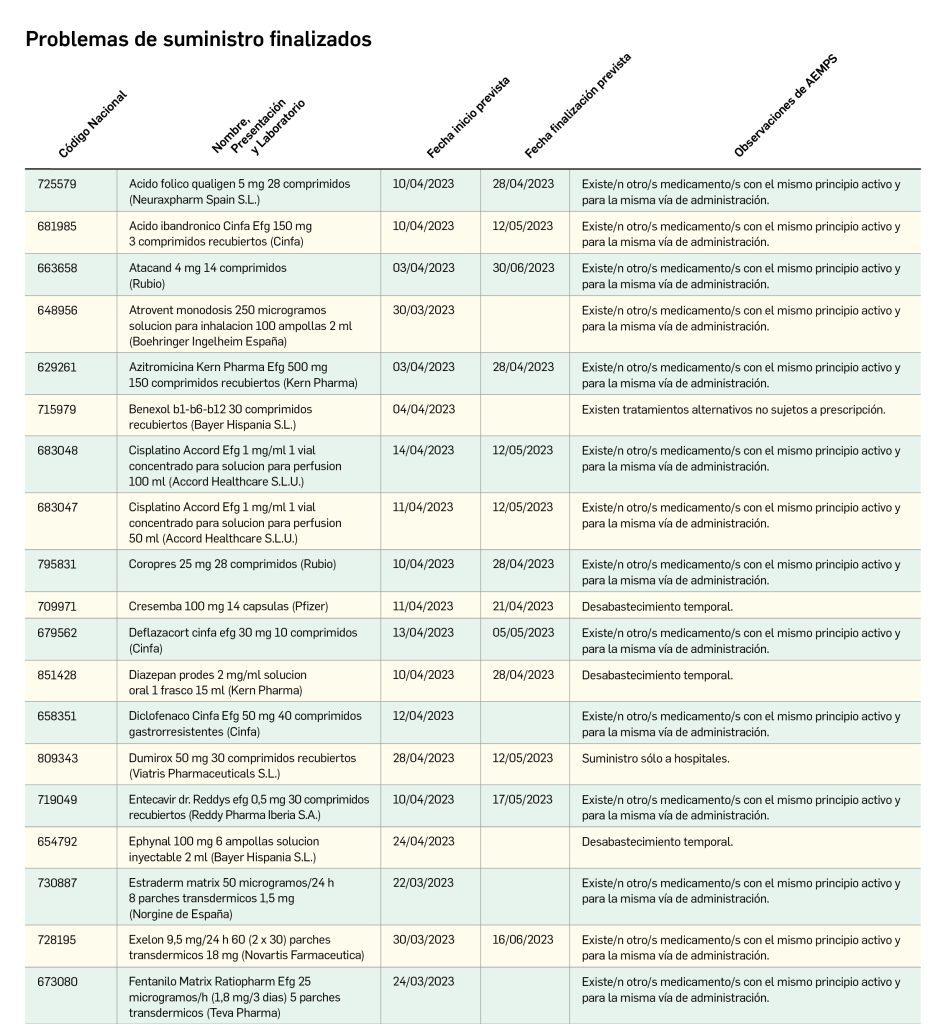
Sin embargo, tal retraso puede quedar mitigado, al menos en parte, por la regulación legal española sobre la disponibilidad de medicamentos en situaciones especiales. En concreto, el Real Decreto 1015/2009, de 19 de junio, regula el acceso a los medicamentos autorizados en otros países (entre ellos los de la Unión Europea) pero no autorizados en España, cuando no cumplan con la definición de uso compasivo de medicamentos en investigación. En su artículo 17 establece que la AEMPS podrá autorizar con carácter excepcional el acceso a dichos medicamentos no autorizados en España y destinados a su utilización en España; su acceso individualizado o cuando se prevea su necesidad para una subpoblación significativa de pacientes, está regulado por los artículos 18 y 19, respectivamente, básicamente a través de las autoridades competentes de las Comunidades Autónomas.
¿Qué medicamentos novedosos?
De los 119 medicamentos autorizados por la Comisión Europea con nuevos principios activos desde el día 1 de enero de 2021 hasta el 30 de marzo de 2023, la mayoría corresponde a antineoplásicos y agentes inmunomoduladores (grupo L de la clasificación ATC), con 44 fármacos (37% del total), entre ellos 6 para cáncer de pulmón, 4 para mieloma múltiple y para esclerosis múltiple, 3 para cáncer de mama, linfoma de célula B, psoriasis, y 2 para melanoma. Les siguen los antiinfecciosos sistémicos (grupo J), con 25 (21%), 14 para COVID-19 y 2 para hepatitis B, infección por VIH y neumonía neumocócica; tracto alimentario y metabolismo (grupo A), con 12 (10%), con 2 para obesidad, prurito asociado colestasis intrahepática y enfermedad de Pompe; sistema nervioso (grupo N), con 8 (7%), con 3 indicados en migraña; y sangre y órganos hematopoyéticos (grupo B), con 7 (6%), 3 de ellos indicados en hemofilia A o B.
Por su parte, de los autorizados por la Comisión Europea, España ha incorporado mayoritariamente los nuevos antiinfecciosos sistémicos (J), con 11 fármacos (34% del total), 9 de ellos indicados para la COVID-19; y antineoplásicos y agentes inmunomoduladores (L), con 10 (31%), con 3 autorizados para la esclerosis múltiple.