Resumen
Abrocitinib es un nuevo inhibidor oral de la Janus cinasa 1 (JAK1), una enzima con actividad tirosina cinasa con múltiples funciones fisiológicas, entre las que se encuentra la transmisión de cascadas de señalización intracelular que modifican la respuesta de las células inmunitarias. Las JAK fosforilan y activan transductores de señales y activadores de la transcripción como STAT, que modifican la expresión génica. La vía de señalización JAK-STAT está estrechamente relacionada con procesos inmunitarios y con trastornos dermatológicos como la dermatitis atópica (DA). En base a los efectos derivados de este mecanismo de acción, abrocitinib ha sido autorizado con indicación en el tratamiento de la dermatitis atópica de moderada a grave en adultos que son candidatos a tratamiento sistémico.
Los datos de eficacia del nuevo fármaco proceden de 5 estudios de fase 3 ya completados y de otro ensayo, también de fase 3, todavía en marcha. Se ha comparado abrocitinib con placebo en tres estudios clínicos, en los cuales el nuevo fármaco mostró superioridad estadística, en mayor medida al utilizar la dosis más elevada (200 mg/día) en términos de IGA 0/1 y de EASI75, consideradas variables coprimarias de eficacia. También se dispone de la comparación con dupilumab, frente al cual abrocitinib parece mostrar una mayor eficacia al inicio del tratamiento, aunque la significación estadística de esta diferencia no ha sido establecida; no obstante, a partir de la semana 16 se ha observado una tendencia a la igualación de la proporción de pacientes con IGA0/1 (en torno al 40-45%) y EASI75 (en torno al 65-70%). En estudios a largo plazo se ha podido establecer la eficacia de abrocitinib en pacientes refractarios a dupilumab.
Los datos de seguridad apuntan a un perfil toxicológico en general similar al de otros inhibidores de JAK. En comparación con placebo, destaca una mayor frecuencia de nasofaringitis (11% vs. 8%), náuseas (10% vs. 2%) y dolor de cabeza (7% vs. 4%). En los estudios clínicos, la incidencia de eventos adversos graves fue baja, incluso en tratamientos de hasta 1 año, siendo el único evento destacable (frecuencia > 1%) las infecciones, aunque menos frecuentes que con placebo (1,4% vs. 1,9%). Sin embargo, existe incertidumbre sobre los posibles efectos de abrocitinib sobre el desarrollo óseo, especialmente en la población pediátrica, lo que ha provocado la restricción de la indicación del fármaco exclusivamente a la población adulta. Tampoco se conocen adecuadamente los efectos a largo plazo respecto al riesgo de neoplasias y otros efectos propios de los inhibidores de JAK, como miopatías (incluyendo rabdomiólisis), perforación gastrointestinal y eventos cardiovasculares graves.
Sin un mecanismo de acción novedoso, en ausencia de datos comparativos directos o indirectos con otros tratamientos sistémicos de uso habitual en el abordaje de la DA de moderada a grave, como ciclosporina A y otros inhibidores de JAK, y teniendo en cuenta las dudas que existen sobre el perfil de seguridad del nuevo fármaco en el tratamiento de esta enfermedad crónica y su restricción a la población adulta, la introducción de abrocitinib no parece que vaya a suponer un cambio sustancial en la terapéutica estándar de la DA.
Aspectos fisiopatológicos
La dermatitis atópica (DA) es una enfermedad inflamatoria de la piel de carácter crónico o crónicamente recurrente caracterizada por la presencia de lesiones eccematosas, sequedad de la piel (xerosis) y prurito intenso. Suele cursar con brotes de duración e intensidad variable, entre los que se intercalan periodos de remisión, aunque, en algunos casos, los síntomas pueden ser continuos. El sistema inmunitario es determinante en la aparición de muchas dermatitis1, y la dermatitis atópica, a diferencia de las dermatitis de contacto, se ha asociado con reacciones de hipersensibilidad tipo I o inmediatas, pues frecuentemente se observa un incremento notable de inmunoglobulinas IgE, como también ocurre en otros trastornos como el asma o la rinitis alérgica (Montalvo-Calvo et al., 2019).
La DA es la manifestación cutánea de un estado denominado atópico o atopia, un término que se define genéricamente como la existencia de hipersensibilidad frente a proteínas heterólogas. La reacción de hipersensibilidad de tipo I se produce tras la exposición a un antígeno exógeno (polen, ácaros, etc.), provocando la liberación inmediata de una amplia variedad de sustancias activas a partir de los mastocitos –también denominados células cebadas– del organismo. Estas sustancias activas, tales como la histamina, prostaglandinas, factor activador de las plaquetas, leucotrienos, factor quimiotáctico de eosinófilos, etc., junto con el aumento de la síntesis de IgE, conforman una situación típica de alergia. La eosinofilia es otro rasgo característico de las reacciones de tipo I.
La etiopatogenia subyacente justifica la notable relación existente entre los pacientes que padecen DA con los antecedentes personales o familiares de crisis asmáticas, rinitis, reacciones cutáneas desproporcionadas tras la picadura de insectos o urticaria masiva. Así, epidemiológicamente, la DA se caracteriza por un historial familiar en el 70% de los pacientes de asma bronquial, rinitis alérgica, fiebre del heno o dermatitis. La probabilidad de padecer la enfermedad aumenta especialmente cuando ambos progenitores la sufrieron; también, aunque en menor medida, cuando únicamente la padeció uno de ellos.
Se trata de una de las enfermedades de la piel más comunes, que puede aparecer en cualquier época de la vida. Se ha estimado que un 15-20% de los niños que acuden al dermatólogo en los países desarrollados padecen este tipo de patología, frente a menos del 1% en los países en vías de desarrollo. La prevalencia en adultos a nivel mundial se cree que oscila entre el 1-3% (Avena-Woods, 2017). Por tanto, se trata de una enfermedad muy ligada al nivel de desarrollo de los países, hasta el punto de que su prevalencia parece haberse triplicado en el último medio siglo en los países desarrollados. En España, se estima que la DA grave tiene una prevalencia del 0,08% de la población. Además, puede afectar a las personas de cualquier raza, es más frecuente en el sexo femenino (1,5:1) y se presenta más a menudo en las clases socioeconómicas altas, así como en las grandes ciudades, posiblemente por una mayor exposición a estímulos capaces de desencadenar el cuadro clínico.
En cuanto a las manifestaciones, el prurito es el síntoma predominante en todos los casos (puede dar lugar a lesiones secundarias debido al rascado). La inflamación epidérmica propia de las dermatitis provoca una serie de lesiones que, progresivamente, según se agrava o se hace crónica, pasan sucesivamente por las fases2 de eritema, edema, vesiculación, exudación, costra, descamación y liquenificación.
No obstante, la presentación y el curso clínico de la DA varían con la edad y suelen ser heterogéneos. En la mayor parte de los pacientes la enfermedad debuta antes de los 5 años, y ésta se resuelve espontáneamente durante la infancia en aproximadamente la mitad de los casos. Es bastante frecuente que la DA aparezca en lactantes a los 3-6 meses de edad con aparición de placas inflamatorias y supuración, o placas con descamación en cara, cuello, superficies de extensión y en la ingle. También es relativamente frecuente su desaparición espontánea entre los 3 y los 5 años de edad, pero puede mantenerse como una condición crónica durante la edad adulta hasta para un 40% de pacientes. Cuando debuta en la edad adulta, la DA suele presentarse como un cuadro eccematoso de aparición alrededor de los 20 años.
Se estima que casi dos tercios (60%) de los pacientes adultos con DA padecen al menos un tipo de comorbilidad atópica, como asma o rinoconjuntivitis alérgica (las tres patologías constituyen la clásica triada de la atopia). También se ha descrito un mayor riesgo de comorbilidades no atópicas, como otros trastornos cutáneos, ciertas enfermedades cardiovasculares, trastornos inmunitarios sistémicos o cáncer. Además, la pérdida de la barrera protectora de la piel y la alteración del sistema inmunitario aumentan el riesgo de infecciones cutáneas, las cuales pueden ser potencialmente graves. Por todo ello, y teniendo en cuenta que las manifestaciones clínicas producen asimismo alteraciones importantes del sueño, secuelas psicológicas y sociales, la DA –sobre todo, en sus formas moderadas y graves– representa un importante problema sociosanitario y tiene un gran impacto en la calidad de vida de los pacientes.
De forma general, la severidad de la DA se determina mediante el empleo de distintas escalas de valoración validadas que consideran la extensión de las áreas de piel afectadas, la gravedad de las lesiones y los síntomas subjetivos del paciente. Las más aceptadas y ampliamente usadas son quizá SCORAD (Scoring Atopic Dermatitis) y EASI (Eczema Area and Severity Index), que no incluyen síntomas subjetivos; según estas escalas se puede definir la DA moderada a partir de una puntuación SCORAD superior a 14 o un EASI desde 7,1 a 21 puntos, y la DA grave como un SCORAD superior a 40 o EASI entre 21,1 y 50 puntos (muy grave: > 50,1-72). Otras escalas muy extendidas son la IGA (Investigator Global Assessment), que valora la enfermedad del 0 al 5 (dermatitis aclarada, mínima, ligera, moderada, intensa y grave) teniendo en cuenta el eritema, la formación de pápulas y de exudados; y la escala NRS (Numerical Rating Scale), escala numérica que mide la intensidad del prurito, siendo 10 la mayor intensidad posible (AEMPS, 2023).
Según se ha sugerido previamente, en la etiopatogenia de la DA no solo intervienen mecanismos inmunitarios, sino también genéticos y ambientales, así como otros característicos de la propia piel del individuo (por ejemplo, se ha postulado el posible papel de anomalías en la sudoración y un aumento de la pérdida transepidérmica de agua), así como factores externos como la dieta, la existencia de aero-alérgenos, infecciones por microorganismos (estafilococos u hongos como Malassezia furfur) o incluso otros factores como sequedad ambiental, disminución de la temperatura, tejidos irritantes, etc., que podrían actuar como estímulos desencadenantes o agravantes de un episodio de DA.
En cualquier caso, en una alta proporción de pacientes con DA –en torno al 80%– se detecta la producción excesiva de IgE y la disminución de la inmunidad mediada por células. De hecho, existe alteración en las subpoblaciones de linfocitos T y de las células de Langerhans (células presentadoras de antígenos existentes en dermis y epidermis): se produce un predominio de células T que contribuye a incrementar la presencia de reacciones inflamatorias, con un descenso asociado de linfocitos CD8+ y un aumento del cociente CD4+/CD8+; además, las células de Langerhans de los pacientes con DA tienen en su superficie una alta expresión del receptor para IgE.
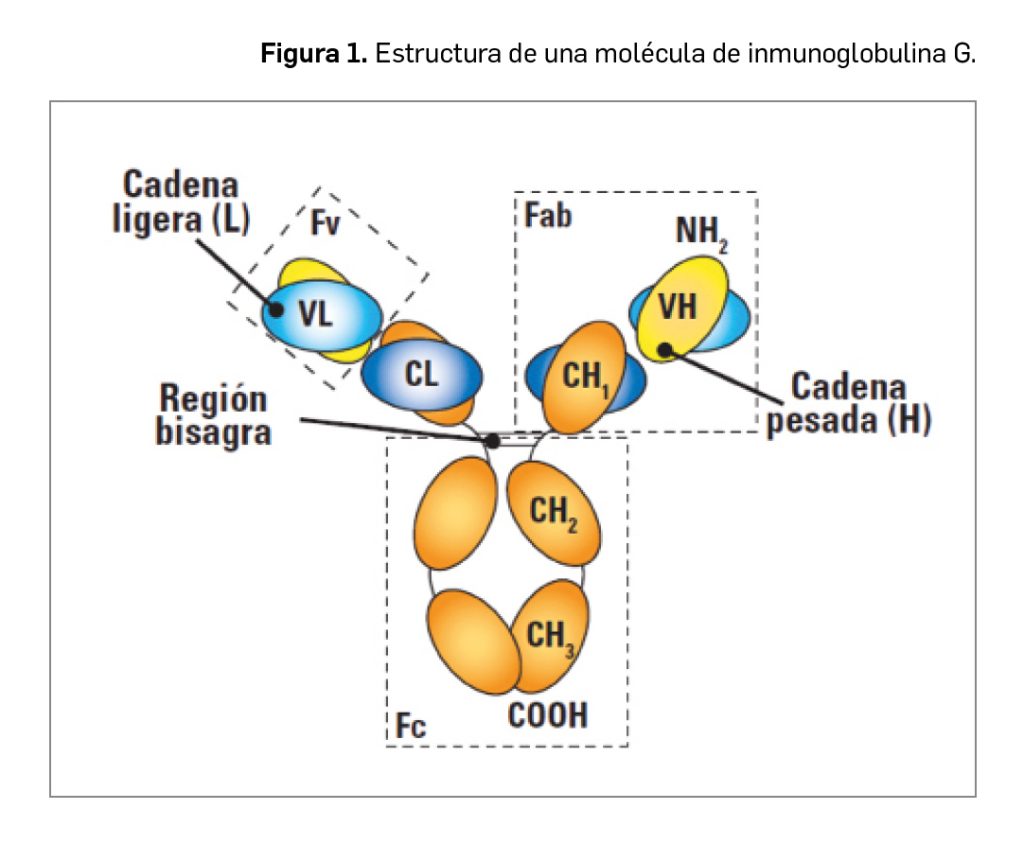
En las lesiones agudas de DA se observa un predominio de linfocitos T facilitadores (Th) de tipo Th2 (subpoblación de CD4+), mientras que en las lesiones liquenificadas y crónicas predominan las células Th1. Los linfocitos Th2 liberan diversos factores (interleucinas IL-4, IL-5, IL-6, IL-10) activadores de las células B, entre los que destaca la sobreproducción de IL-4, que estimula la producción de IgE e inhibe la de interferón gamma (IFN-γ), reduciendo la diferenciación de células T hacia Th1. Por su parte, las células Th1 median la producción de IL-2, IFN-γ y factor de necrosis tumoral (TNF), activando los macrófagos y favoreciendo la reacción de hipersensibilidad retardada. Por tanto, son las células Th2 las responsables de la producción aumentada de IL-4 e IL-13, estimuladoras de la producción de IgE, IL-5 (responsable de la activación de los eosinófilos) e IL-10 (responsable de la disminución de la inmunidad celular). Estos hallazgos son importantes ya que la IL-4 y la IL-13 inducen la expresión de moléculas de adhesión involucradas en la migración de células inflamatorias en las zonas de inflamación tisular (Figura 1).
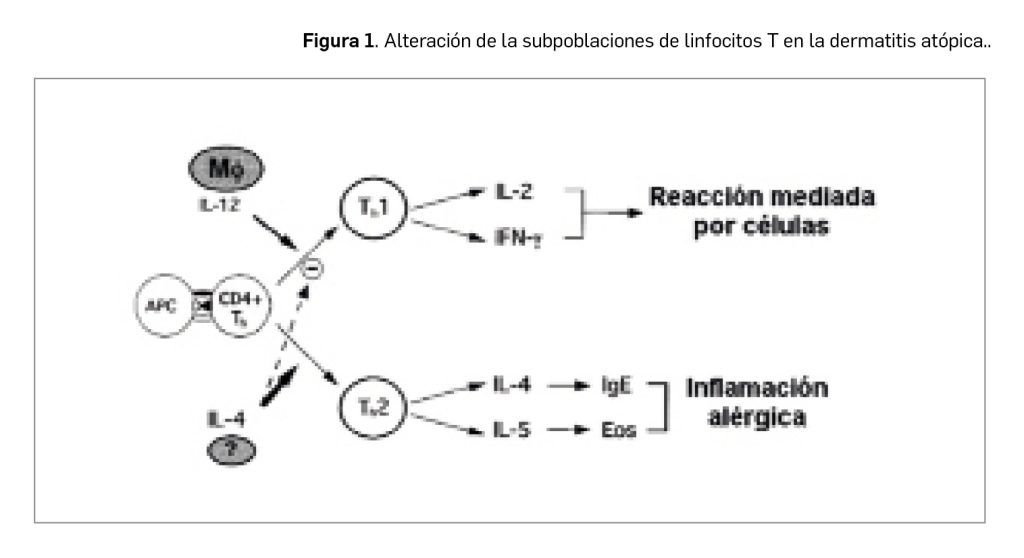
Este conjunto de alteraciones inmunitarias condicionan, en consecuencia, la aparición frecuente de infecciones cutáneas recidivantes, tanto de origen bacteriano como vírico o fúngico. De ellas, quizás la más frecuente sea la colonización por Staphylococcus aureus, presente en el 90% de las lesiones cutáneas.
En base a todo lo anterior, los objetivos fundamentales del tratamiento de la dermatitis atópica consisten en limitar los síntomas (sobre todo, el prurito), prevenir las exacerbaciones, evitar las infecciones dérmicas y minimizar los riesgos del tratamiento. Para ello, previamente a la farmacoterapia, en esta patología es fundamental la adopción de medidas de higiene a fin de prevenir todas aquellas circunstancias que desencadenen o intensifiquen el prurito. Entre otras, será recomendable administrar cremas emolientes, evitar la temperatura elevada y una baja humedad ambiental (la calefacción por corrientes de aire caliente puede ser perjudicial), así como el uso de ropas de abrigo excesivas y el contacto con ciertos tejidos (lana, plásticos y gomas); por el contrario, el uso de guantes y la exposición solar suelen resultar beneficiosas. En cuadros de carácter crónico, es recomendable el empleo frecuente de soluciones oleosas que limpien y humedezcan la piel.
La farmacoterapia estándar frente a los síntomas de la DA se basa en la administración de agentes antiinflamatorios tópicos, sobre todo, corticosteroides (beclometasona, betametasona, fluocinolona, metilprednisolona, etc.) e inhibidores de la calcineurina (tacrolimus o pimecrolimus) en forma de cremas; sin embargo, las preparaciones tópicas producen resultados pobres (sobre todo en casos moderados-graves), posiblemente debido a su escasa penetración cutánea. Junto a estos, suelen administrarse emolientes e hidratantes de la piel, tales como vaselina blanca, vaselina hidrófila, óxido de zinc, etc. La utilización de corticosteroides sistémicos en ciclos cortos se reserva para los casos agudos o de gran extensión corporal. Además, en caso de infección cutánea sobreañadida se usan antibióticos por vía sistémica.
No es aconsejable la utilización por vía tópica de antihistamínicos, neomicina, sulfamidas ni perfumes. El tratamiento del picor asociado a la dermatitis se realiza generalmente mediante la utilización de antihistamínicos por vía oral: suele preferirse a los antiguos antihistamínicos (por ejemplo, dexclorfeniramina), que producen más somnolencia que los modernos, ya que aparentemente su eficacia se debe más a su efecto sedante que a una actividad antipruriginosa propiamente dicha. Cuando en la epidermis predomina el edema y la exudación, es útil el empleo tópico de sulfato de cobre o permanganato potásico por sus propiedades astringentes y desinfectantes.
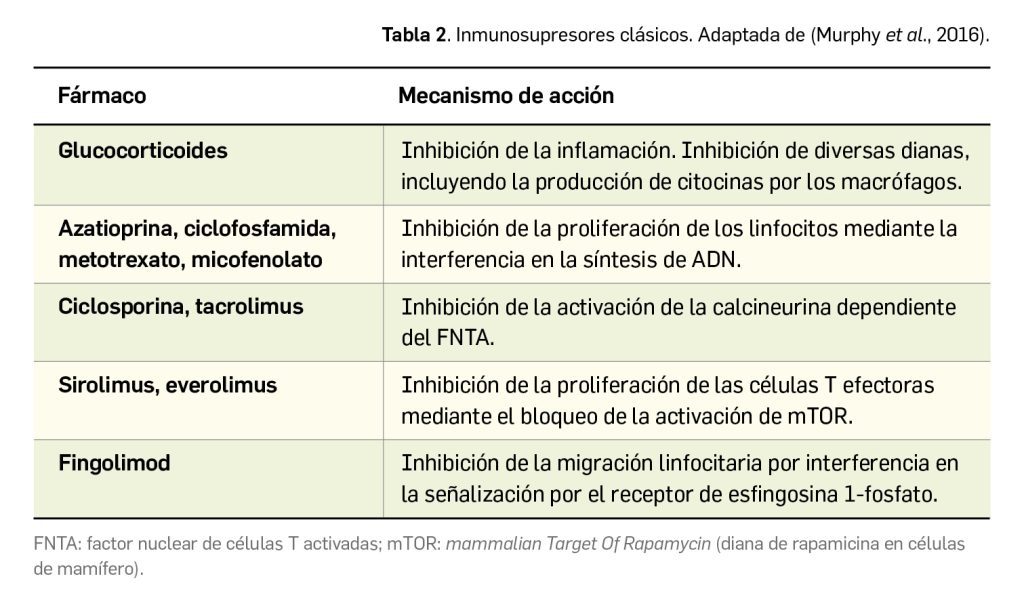
Los pacientes con enfermedad grave o resistente a los tratamientos tópicos pueden requerir de fototerapia (UVA) o ser candidatos para la administración de tratamiento sistémico con inmunosupresores convencionales: el único autorizado en la UE, y considerado de elección, es la ciclosporina A, aunque también se emplean –con menor evidencia que respalde su uso– corticoides, azatioprina, metotrexato y micofenolato de mofetilo. La decisión de iniciar tratamiento sistémico en los pacientes con DA moderada-grave se debe de basar en la evaluación de la severidad y la calidad de vida y, al mismo tiempo, en la consideración del estado general de salud de forma individualizada para cada paciente, valorando factores como preferencias del paciente, comorbilidades y coste del tratamiento.
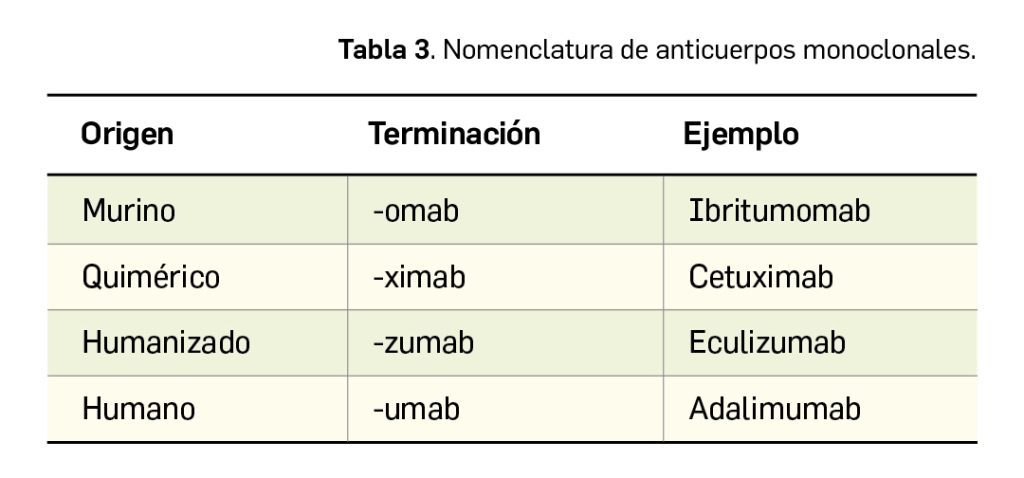
Dentro del arsenal para el tratamiento sistémico se han producido avances en los últimos años con la introducción de anticuerpos monoclonales e inhibidores selectivos de cinasas Janus. Entre los primeros sobresale dupilumab, un monoclonal que se dirige de forma específica a la subunidad α del receptor de la interleucina 4 (IL-4Rα), impidiendo así la señalización molecular mediada por la unión de esa citocina a su receptor tipo I y también la señalización de IL-4 e IL-13 a través del receptor tipo II. Dado que IL-4 e IL-13 son los principales mediadores de la inflamación de tipo 2, dupilumab ejerce efectos antiinflamatorios de utilidad en enfermedades relacionadas con la atopia. También se encuentra aprobado en DA moderada-grave tralokinumab, un monoclonal dirigido específicamente a un epítopo de la IL-13, que previene la unión de esta citocina tanto a los receptores IL-13Rα1/IL-4Rα como a los IL-13Rα2, y así neutraliza la actividad biológica de la IL-13.
Por otro lado, los inhibidores de cinasas Janus (JAK, del inglés Janus kinases) actúan como inmunosupresores. Las JAK cinasas son una familia de enzimas de las que hasta ahora se conocen cuatro miembros (JAK1, JAK2, JAK3 y TYK2), expresadas en prácticamente todas las células del organismo (con excepción de JAK3, que se restringe a células hematopoyéticas) y con múltiples funciones fisiológicas. Ciertas mutaciones en estas enzimas o fallos en su cascada de señalización intracelular se han relacionado con trastornos mieloproliferativos, autoinmunes e inflamatorios, incluyendo la DA. Baricitinib es un inhibidor selectivo y reversible de JAK1 y JAK2, mientras que upadacitinib es un inhibidor selectivo y reversible de JAK1 y de JAK1/3. Ambos se encuentran aprobados con indicación en DA moderada-grave en pacientes candidatos a tratamiento sistémico.
A pesar de la incorporación de estos nuevos tratamientos, el abordaje de las más severas de la DA continúa siendo actualmente un reto terapéutico y una necesidad médica no resuelta. Las limitaciones existentes en el empleo a largo plazo de ciclosporina A como consecuencia de su toxicidad, así como la elevada proporción de pacientes que son refractarios a otros tratamientos sistémicos, son factores que hacen necesaria la ampliación de las alternativas terapéuticas.
Acción y mecanismo
Abrocitinib es un nuevo inhibidor de la Janus cinasa 1 (JAK1), una tirosina cinasa con múltiples funciones fisiológicas, entre las que se encuentra la transmisión de cascadas de señalización intracelular que modifican la respuesta de las células inmunitarias. Las JAK fosforilan y activan transductores de señales y activadores de la transcripción como STAT, que actúa modificando la expresión génica. La vía de señalización JAK-STAT se encuentra estrechamente relacionada con procesos inmunitarios y con trastornos dermatológicos. En base a los efectos derivados de este mecanismo de acción, abrocitinib ha sido autorizado con indicación en el tratamiento de la dermatitis atópica de moderada a grave en adultos que son candidatos a tratamiento sistémico.
En estudios in vitro se ha podido demostrar una inhibición selectiva de JAK1 por parte de abrocitinib, aunque en un contexto celular la inhibición de JAK1 puede afectar a otras vías de señalización mediadas por otros miembros de la familia JAK (EMA, 2021). También en estudios preclínicos, en este caso en cultivos de células humanas, se observó que abrocitinib tenía mayor afinidad por dímeros de señalización en los que estaba presente JAK1 en comparación con aquellos dímeros en los que no se encontraba esta proteína, tales como JAK2/TYK2 o JAK2/JAK2.
Aspectos moleculares
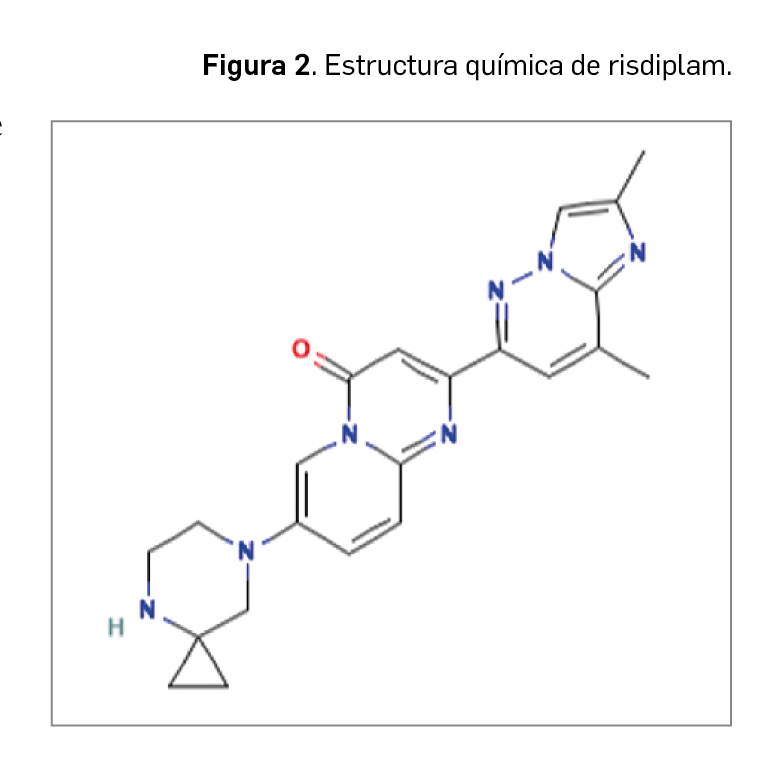
Abrocitinib es, desde el punto de vista químico, el N-((1S,3S)-3-(metil(7H-pirrolo[2,3-d]pirimidin-4-il)amino)ciclobutil)propano-1-sulfonamida. Se corresponde con la fórmula molecular C14H21N5O2S y tiene una masa molecular de 323,42 D.
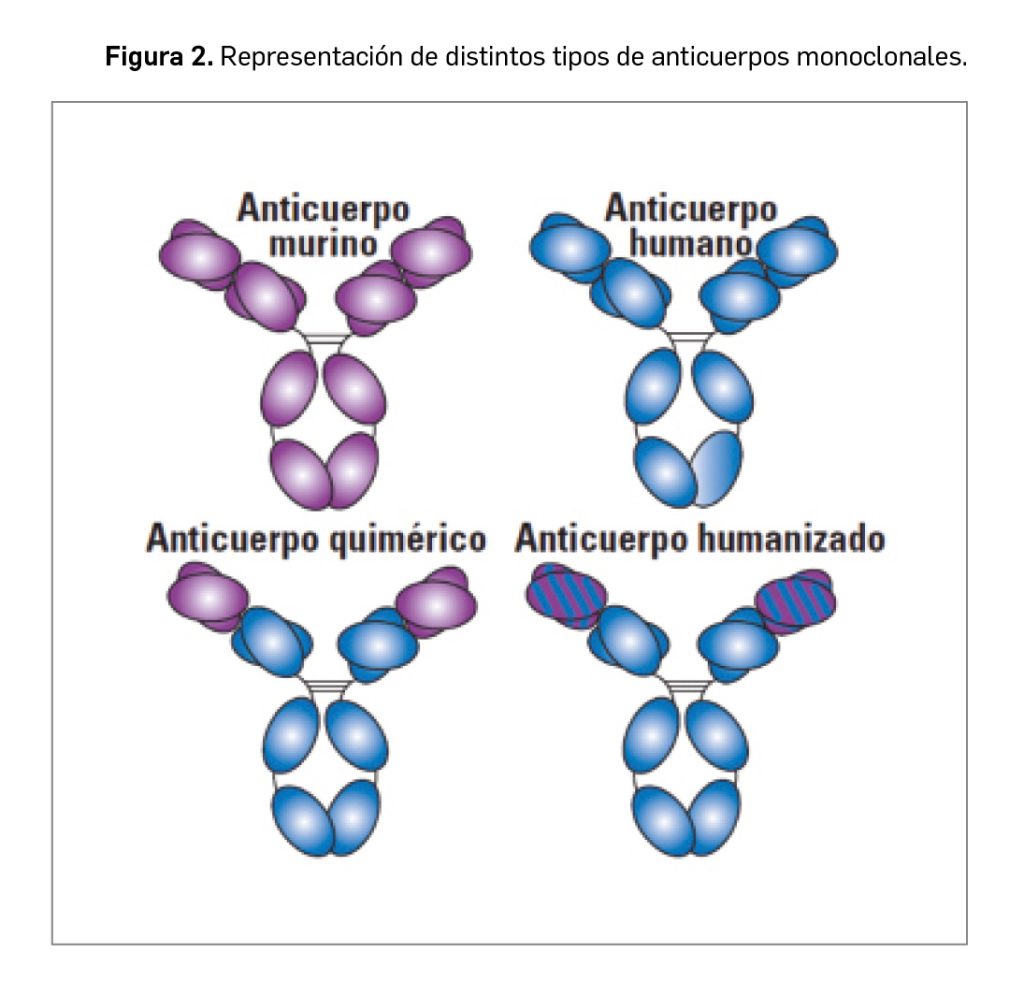
Como se puede observar en la Figura 2, abrocitinib guarda una estrecha similitud estructural con otros inhibidores de JAK empleados en DA como baricitinib y upadacitinib, especialmente con el primero. Abrocitinib y baricitinib presentan un anillo de pirimidina fusionado a un anillo de pirrol, así como un grupo sulfona unido a un grupo etilo.
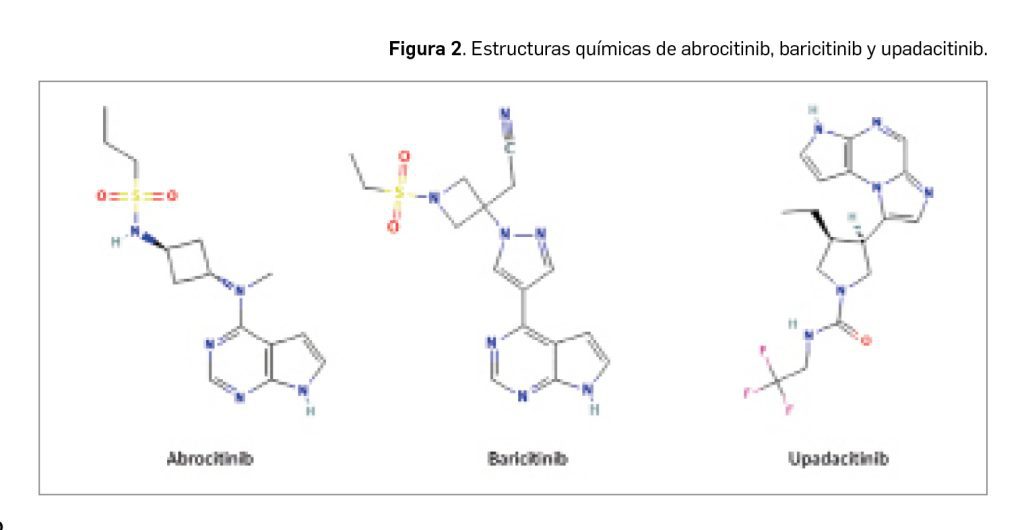
En forma purificada, abrocitinib se presenta como un polvo cristalino blanco a violeta pálido o rosa pálido, escasamente soluble.
Abrocitinib se une de forma preferente a la cinasa JAK1 a través de enlaces de hidrógeno con los residuos aminoacídicos Glu957, Leu959 y Asn1008. También se establecen interacciones hidrofóbicas entre los anillos de pirrol y pirimidina y varios aminoácidos de la enzima y enlaces hidrógeno-carbono con los residuos Leu88 y Leu959 (Shawky et al., 2022).
Eficacia y seguridad clínicas
La eficacia y seguridad clínicas del tratamiento por vía oral con abrocitinib en su pauta autorizada han sido adecuadamente contrastadas en hasta cinco estudios clínicos de fase 3 y continúan siendo evaluadas en un estudio de extensión a largo plazo todavía en marcha.
Los estudios pivotales Mono-1 y Mono-2, con idéntico diseño, fueron dos estudios aleatorizados, doble ciego, controlados por placebo, de grupos paralelos y multicéntricos en los que se evaluó la eficacia y la seguridad de abrocitinib en monoterapia, respectivamente, en un total de 387 y 391 pacientes de 12 o más años con DA de moderada a severa. Las variables coprimarias de eficacia de estos estudios fueron la respuesta al tratamiento en la escala IGA (0/1 y una reducción respecto al inicio de ≥ 2 puntos) en la semana 12 y la respuesta EASI75 o superior respecto a la línea de base en la semana 12; como variables secundarias se consideraron la mejora del prurito (de ≥ 4 puntos en la escala PP-NRS4 en las semanas 2, 4 y 12) y el cambio respecto a la línea de base en la escala PSAAD3 en la semana 12.
En cuanto a las características basales de los participantes, bien balanceadas entre los grupos de tratamiento, destaca que aproximadamente un 42% fueron mujeres, con mayoría de raza caucásica (59-72%), y la edad mediana se situó entre 29 y 31 años. La duración mediana de la enfermedad fue de alrededor de 20 años y, en cuanto a las características clínicas basales de la enfermedad, en las distintas variables de análisis se mantuvo una adecuada proporcionalidad entre ambos estudios.
Los resultados divulgados reflejan la superioridad de abrocitinib frente a placebo en el control de la DA, con una mayor eficacia de la dosis de 200 mg/día frente a la dosis diaria de 100 mg. Los principales resultados de eficacia de los estudios Mono-1 (Simpson et al., 2020) y Mono-2 (Silverberg et al., 2020) respecto a las variables coprimarias se sintetizan en la Tabla 1 y en la Tabla 2. En cuanto a las variables secundarias, la mejora del prurito en la semana 12 (así como en los puntos intermedios de análisis) también fue significativamente superior con el tratamiento con abrocitinib, tanto en la escala PP-NRS4 como en PSAAD (p < 0,001).
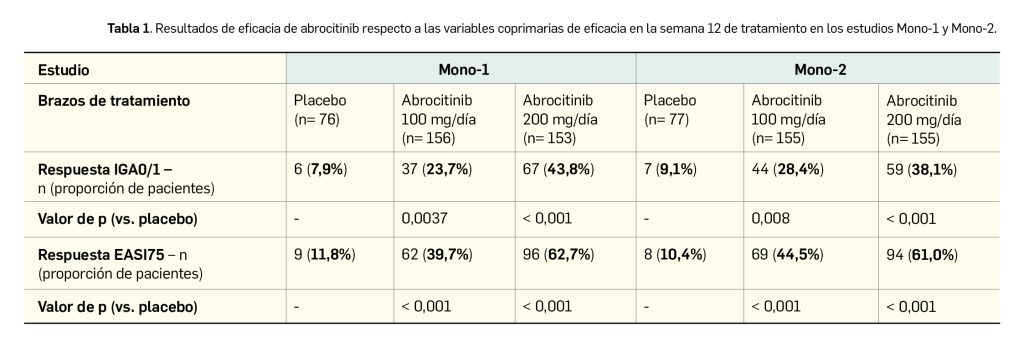
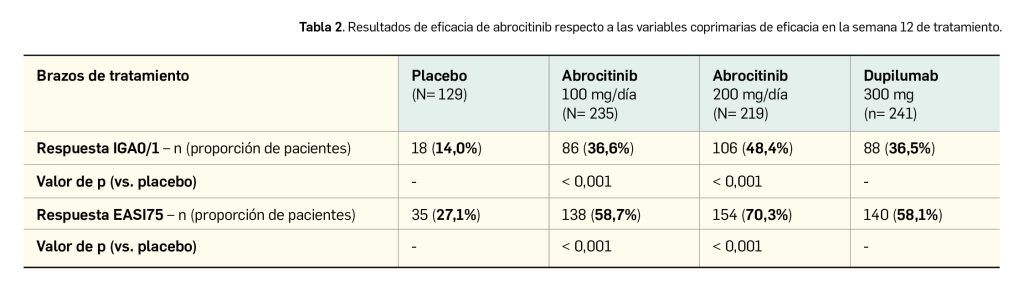
Por otro lado, se dispone también de los resultados del estudio COMPARE, otro ensayo de fase 3 aleatorizado, doblemente ciego, controlado por placebo, de grupos paralelos y multicéntrico en el que se evaluó la eficacia de abrocitinib y de dupilumab en comparación con placebo en 838 pacientes adultos con DA de moderada a severa que recibían tratamiento tópico de forma concomitante. Las variables coprimarias de eficacia fueron las mismas que en los estudios Mono-1 y Mono-2, mientras que la respuesta IGA 0/1 y EASI75 en la semana 16 fueron variables secundarias. Alrededor de tres cuartas partes de los pacientes (72%) eran caucásicos, con mayoría de mujeres (51%) y una edad mediana de 34 años. Las características basales de la enfermedad estuvieron adecuadamente balanceadas entre los distintos brazos de tratamiento.
Los principales resultados de eficacia del estudio COMPARE (Bieber et al., 2021) se presentan en la Tabla 2. Un tratamiento de 12 semanas con abrocitinib se mostró significativamente superior a placebo en términos de las variables coprimarias de eficacia; los resultados también parecen favorables al nuevo fármaco frente a dupilumab si se considera su dosis más elevada, aunque no se dispone de un contraste de hipótesis para las variables coprimarias entre ambos principios activos, por lo que no se puede concluir sobre la significación de este resultado.
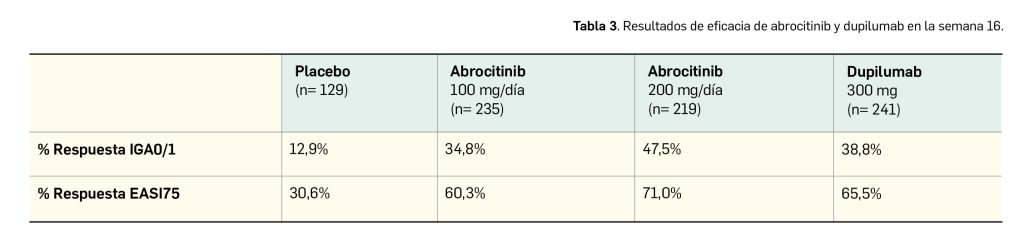
No obstante, a la semana 16 (Tabla 3), los datos de eficacia reflejan una tendencia favorable para dupilumab frente a la dosis inferior de abrocitinib y una menor diferencia absoluta respecto de la dosis superior en comparación con la semana 12.
El mantenimiento a más largo plazo de la eficacia de abrocitinib se ha evaluado en el estudio REGIMEN, un ensayo de fase 3 de inducción con retirada aleatorizada, doble ciego, controlado por placebo y multicéntrico en el que participaron 1233 pacientes de 12 o más años con DA de moderada a severa.
De acuerdo a los resultados publicados (Blauvelt et al., 2022), del total de pacientes incluidos, en 798 (64,7%) se obtuvo respuesta a la inducción con abrocitinib 200 mg/día durante 12 semanas. Estos pacientes respondedores fueron aleatorizados posteriormente para recibir abrocitinib 100 mg/día (n= 265), abrocitinib 200 mg/día (n= 266) o placebo (n= 267) durante un periodo de mantenimiento de 40 semanas. En esa fase, la dosis más elevada de abrocitinib resultó la más eficaz en la prevención de un brote de DA, que apareció en un 16,5% de los participantes, frente al 39,6% de los tratados con la dosis de 100 mg/día y el 77,5% de aquellos que recibieron placebo hasta la semana 40. La reducción del riesgo de la dosis de 200 mg/día frente a placebo fue estadísticamente significativa (p < 0,0001).
Aquellos pacientes en quienes se observó una pérdida de respuesta o que sufrieron un brote (n= 351) entraron en una fase de rescate con abrocitinib 200 mg/día junto con medicaciones tópicas durante 12 semanas (AEMPS, 2023), con respuestas IGA 0/1 y EASI75 superiores en los pacientes previamente tratados con placebo (81,6% y 91,8%, respectivamente) y con abrocitinib 100 mg/día (58,8% y 74,5%, respectivamente) frente a quienes habían tenido pérdida de respuesta con la dosis de abrocitinib de 200 mg/día (36,6% y 55,0%).
Se dispone también de una comparación de abrocitinib frente a dupilumab en el estudio DARE (Reich et al., 2022), otro fase 3, aleatorizado, doble ciego y multicéntrico en el que participaron pacientes adultos con DA de moderada a grave. A pesar de que en las semanas iniciales de tratamiento se observó una mayor proporción de pacientes con respuesta (EASI90) en el brazo de abrocitinib, las diferencias tendieron a disminuir con el tiempo, siendo en la semana 16 del 54% vs. 42% a favor de abrocitinib, mientras que en la semana 26 no hubo diferencias observables (AEMPS, 2023).
Por último, en el estudio EXTEND, todavía en marcha y en el que se evalúan la eficacia y seguridad de abrocitinib a largo plazo en pacientes de 12 o más años con DA de moderada a grave. Se trata de un estudio aleatorizado de fase 3 en que participan 3163 pacientes que habían completado alguno de los estudios anteriores. El estudio consiste en una primera fase ciega de 92 semanas de duración, seguida de una fase abierta de duración variable. Los resultados hasta ahora disponibles revelan una respuesta a las 12 semanas en pacientes que previamente habían fracasado al tratamiento con dupilumab, mayor con la dosis de 200 mg/día de abrocitinib (IGA 0/1: 47%; EASI75: 80%).
En cuanto a la seguridad de abrocitinib, se dispone de datos agregados provenientes de los estudios en los que el fármaco se comparó con placebo, con más de 1500 pacientes tratados. La incidencia de eventos adversos fue mayor con abrocitinib (63% vs. 54% con placebo), destacando una mayor incidencia de infecciones (35% vs. 26%), de molestias gastrointestinales (18% vs. 8%) y de trastornos del sistema nervioso (11% vs. 6%); en cambio, las afecciones de la piel fueron más comunes en los pacientes tratados con placebo (15% vs. 17%). En concreto, las reacciones adversas más comunes fueron nasofaringitis (11% vs. 8%), náuseas (10% vs. 2%) y dolor de cabeza (7% vs. 4%), no identificándose un perfil toxicológico distinto entre adultos y pacientes menores de 18 años.
Cabe citar que se reportaron 3 fallecimientos durante el tratamiento y seguimiento, todos en pacientes tratados con abrocitinib, aunque ninguna muerte se consideró relacionada con el tratamiento. Los eventos adversos graves en tratamientos de hasta 16 semanas fueron poco comunes tanto en quienes recibieron el nuevo fármaco como en los que recibieron placebo (2,5% vs. 3,2%). En tratamientos más prolongados, únicamente se observó una incidencia superior al 1% de eventos graves en el caso de infecciones (1,4% para abrocitinib vs. 1,9% para placebo). La EMA (2021) consideró como eventos adversos de especial interés el tromboembolismo venoso, las infecciones graves (incluyendo infecciones por herpes zóster) y la aparición de neoplasias, entre otros. En el caso de la población adolescente, existe incertidumbre sobre el posible efecto del tratamiento con abrocitinib sobre el desarrollo óseo.
Aspectos innovadores
Abrocitinib es un nuevo inhibidor de la Janus cinasa 1 (JAK1), una enzima con actividad tirosina cinasa con múltiples funciones fisiológicas, entre las que se encuentra la transmisión de cascadas de señalización intracelular que modifican la respuesta de las células inmunitarias. Las JAK fosforilan y activan transductores de señales y activadores de la transcripción como STAT, que actúa modificando la expresión génica: esta vía de señalización JAK-STAT se encuentra estrechamente relacionada con procesos inmunitarios y con trastornos dermatológicos. En base a su mecanismo de acción, abrocitinib ha sido autorizado con indicación en el tratamiento de la dermatitis atópica (DA) de moderada a grave en adultos que son candidatos a tratamiento sistémico.
La evaluación de la eficacia y la seguridad clínicas de abrocitinib ha sido considerada como adecuada por las agencias reguladoras, comprendiendo hasta 5 estudios de fase 3 ya finalizados y otro estudio de extensión de fase 3 todavía en marcha en pacientes con DA de moderada a grave.
Dos de ellos (Mono-1 y Mono-2), con idéntico diseño, examinaron la eficacia del nuevo fármaco en monoterapia y en comparación con placebo, con resultados estadísticamente significativos favorables a la dosis más alta de abrocitinib (200 mg/día) tras 12 semanas de tratamiento en cuanto a la proporción de pacientes que alcanzó los objetivos coprimarios de IGA0/1 y EASI75. En estos estudios, la proporción de respuesta IGA0/1 con abrocitinib 200 mg fue del 38%-44%% vs. 8%-9% con placebo y la de EASI75 fue del 61%-63% vs. 10%-12%.
En el estudio REGIMEN, también en comparación con placebo, abrocitinib se mostró superior en eficacia a más largo plazo (52 semanas sumando las fases de inducción y mantenimiento) en términos de prevención de un brote de DA, pasando de un 77,5% de pacientes que sufrieron un brote tras recibir placebo frente a un 16,5% de los que recibieron abrocitinib 200 mg. Sus resultados indican una reducción estadísticamente significativa frente a placebo (p < 0,0001) del riesgo de brote con la dosis alta en los pacientes con respuesta a abrocitinib en la fase de inducción (65%).
Por otro lado, el estudio COMPARE evaluó comparativamente la eficacia de abrocitinib frente a dupilumab. En la semana 12 de tratamiento, el tratamiento con el nuevo fármaco a la dosis más elevada resultó en mejores datos respecto a dupilumab en términos de IGA 0/1 (48% vs. 37%) y de EASI75 (70% vs. 58%), aunque no se evaluó la significación estadística de este resultado. Sin embargo, en la semana 16 los resultados de eficacia de dupilumab continuaron mejorando, mientras que los de abrocitinib se mantuvieron estables, lo cual puede ser consecuencia de que el tratamiento con dupilumab presenta un mayor periodo de latencia que retrasa el tiempo hasta que se alcanza un efecto terapéutico completo (AEMPS, 2023). En este sentido, los resultados del estudio DARE también apuntan a que en tratamientos más prolongados (de hasta medio año), la eficacia de ambos tratamientos sería similar.
Los datos con mayor seguimiento actualmente disponibles proceden del estudio EXTEND, todavía en marcha, que sugieren que en pacientes refractarios al tratamiento con dupilumab es posible obtener respuestas clínicas con abrocitinib, especialmente al emplear la dosis diaria de 200 mg (IGA0/1 de 47% y EASI75 del 80% en pacientes con respuesta).
Desde el punto de vista de la seguridad, el perfil toxicológico del nuevo fármaco parece clínicamente manejable y consistente con lo conocido para otros inhibidores de JAK, destacando la aparición de eventos adversos de tipo gastrointestinal, infecciosos y del sistema nervioso; en comparación con placebo, se vio una mayor frecuencia de nasofaringitis (11% vs. 8%), náuseas (10% vs. 2%) y dolor de cabeza (7% vs. 4%). La incidencia de eventos adversos graves fue baja, incluso en tratamientos de 1 año de duración, siendo las infecciones el único evento notificado en más del 1% de pacientes, menos frecuente con abrocitinib que con placebo (1,4% vs. 1,9%). No obstante, persisten ciertas incertidumbres sobre la seguridad del fármaco, relativas a su posible influencia sobre el desarrollo óseo (en población pediátrica) y a sus efectos a largo plazo sobre el riesgo de neoplasias y otros eventos asociados a este tipo de fármacos, como miopatías o eventos cardiovasculares graves.
Debido a estos motivos, la EMA ha emitido una opinión positiva para la autorización de comercialización de abrocitinib, a la espera de que el TAC aporte datos procedentes de los informes periódicos actualizados de seguridad y desarrolle un plan de minimización de riesgos.
Abrocitinib no ha sido comparado por ahora de forma directa o indirecta con otros inhibidores de JAK empleados en DA (baricitinib, upadacitinib) ni con ciclosporina A, considerado el estándar de tratamiento de las formas moderadas o graves de la enfermedad, lo que dificulta determinar el posicionamiento del nuevo fármaco respecto a sus alternativas. Además, la comparación con dupilumab no arrojó grandes diferencias entre ambos tratamientos, aunque los datos a más largo plazo sugieren que abrocitinib podría ser útil en pacientes refractarios a dupilumab. La seguridad del nuevo fármaco en tratamientos a largo plazo es un aspecto sobre el que se requiere información adicional, a pesar de que los datos disponibles apuntan a un perfil similar al de otros inhibidores de JAK. Por tanto, de acuerdo a los resultados de eficacia y seguridad obtenidos en los ensayos clínicos, no parece que abrocitinib vaya a implicar un cambio relevante en el manejo de la DA, sin tampoco incorporar un mecanismo de acción novedoso en la indicación.