Resumen
Bedaquilina es un nuevo antibiótico oral con actividad frente a la tuberculosis multirresistente, que actúa inhibiendo de manera específica a la enzima ATP sintasa micobacteriana, esencial en la producción de energía para el microorganismo: produce así un efecto bactericida tanto en las bacterias en reproducción como en las bacterias en reposo (fase latente). Se encuentra actualmente autorizado y está comercializado como medicamento huérfano, estando indicado en pacientes adultos y pediátricos (de 5 a menos de 18 años y un peso de al menos 15 kg) para ser utilizado como parte de un adecuado tratamiento combinado de la tuberculosis pulmonar multirresistente (MDR-TB) cuando un régimen de tratamiento efectivo no puede instaurarse por motivos de resistencia o tolerabilidad.
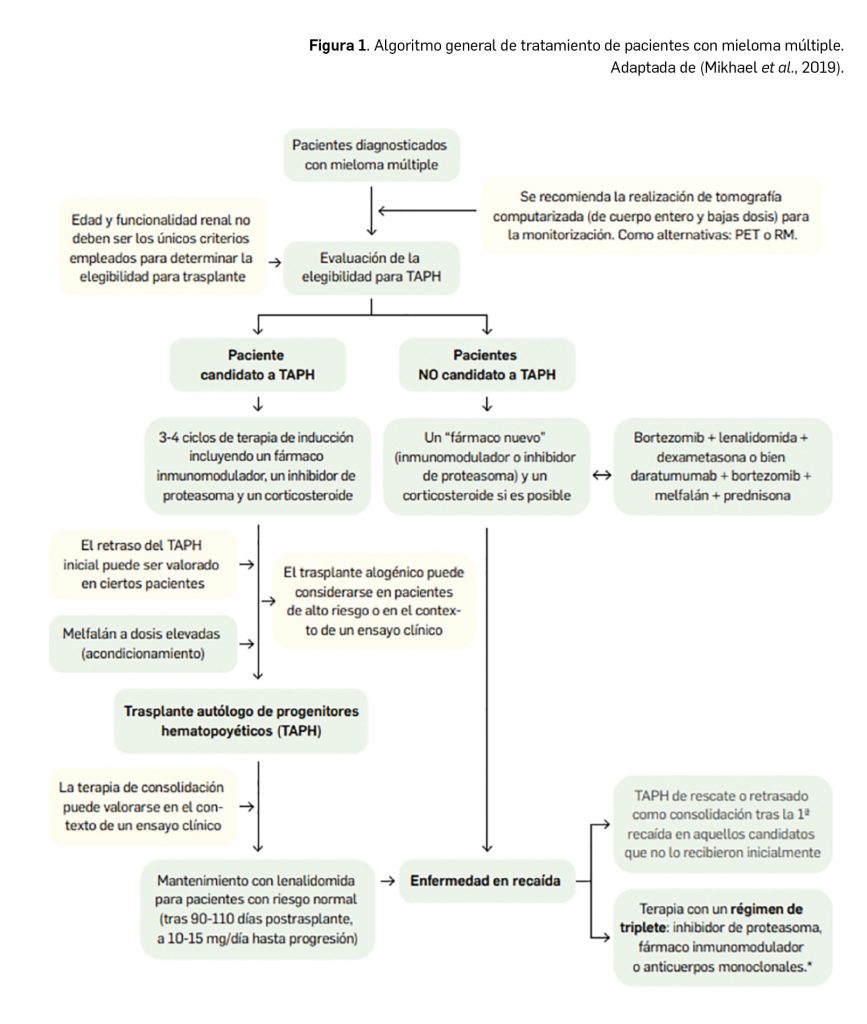
Se dispone de los datos de eficacia de dos estudios de fase 2 en pacientes adultos con MDR-TB que han apoyado la autorización del fármaco en la UE. El ensayo pivotal C208 fue un estudio controlado por placebo, doble ciego y aleatorizado, cuyos resultados indican una reducción estadísticamente significativa de 42 días en el tiempo mediano hasta la conversión del cultivo de esputo en pacientes tratados con un régimen que incluyó bedaquilina (83 vs. 125 días). En el ensayo C209, de fase 2b y de un solo brazo, con la misma variable de eficacia, se obtuvo un tiempo mediano hasta la negativización del cultivo de esputo de 57 días, con una estrategia terapéutica más individualizada, semejante a la práctica clínica habitual. Más recientemente se han publicado los resultados de un ensayo de fase 3 que indican la superioridad del tratamiento combinado con bedaquilina en comparación con un régimen sin este fármaco en relación a la tasa de respuesta (83% vs. 71%; p< 0,0001). Los estudios que sustentan la indicación pediátrica se encuentran todavía en marcha, pero se ha considerado que la eficacia y seguridad de bedaquilina son extrapolables a partir de los resultados obtenidos en población adulta, con el debido ajuste de dosis.
Respecto a la seguridad del nuevo fármaco, durante los estudios clínicos de fase 2 se observó una elevada proporción de eventos adversos, cercana al 100%, aunque similar en el brazo de tratamiento y en el brazo de placebo. Los eventos adversos reportados en mayor medida con bedaquilina fueron artralgias, dolor de cabeza y hemoptisis. Los eventos adversos graves fueron poco frecuentes y, aunque ligeramente más incidentes con bedaquilina (7% vs. 2%), ninguno tuvo una frecuencia superior al 1%. En su perfil toxicológico preocupa potencial arritmogénico (capacidad para alargar el intervalo QT) y una potencial hepatotoxicidad, que requieren de monitorización en estudios de fase 3 para una mejor caracterización del beneficio-riesgo.
En definitiva, se trata de la primera arilquinolina aprobada para el tratamiento de la tuberculosis multirresistente, con un innovador mecanismo de acción en la indicación (inhibición de la ATP sintasa de M. tuberculosis). Su inclusión en un régimen combinado de tratamiento, especialmente junto con un aminoglucósido inyectable y/o una fluoroquinolona, permitirá incrementar la eficacia en la erradicación de las micobacterias, además de relacionarse con un perfil de seguridad aceptable, a pesar de las incertidumbres en cuanto a su potencial arritmogénico y su hepatotoxicidad. En el contexto de un tratamiento complejo y de larga duración en el que la aparición de resistencias intra-tratamiento es frecuente, bedaquilina puede contribuir a afrontar una necesidad médica no cubierta, pero la limitación de su indicación a la ausencia de alternativas impide por ahora considerarlo como un progreso disruptivo en el tratamiento de la tuberculosis multirresistente.
Aspectos fisiopatológicos
La tuberculosis es una enfermedad infecciosa provocada por el bacilo Mycobacterium tuberculosis, una bacteria intracelular que produce una infección respiratoria. Aunque se estima que aproximadamente una tercera parte de la población mundial se encuentra infectada por la bacteria, solo una pequeña parte de las personas desarrolla la enfermedad (5-10% de los infectados), generalmente como consecuencia de una disminución de la competencia del sistema inmunitario, en muchos casos relacionada con la infección por el VIH.
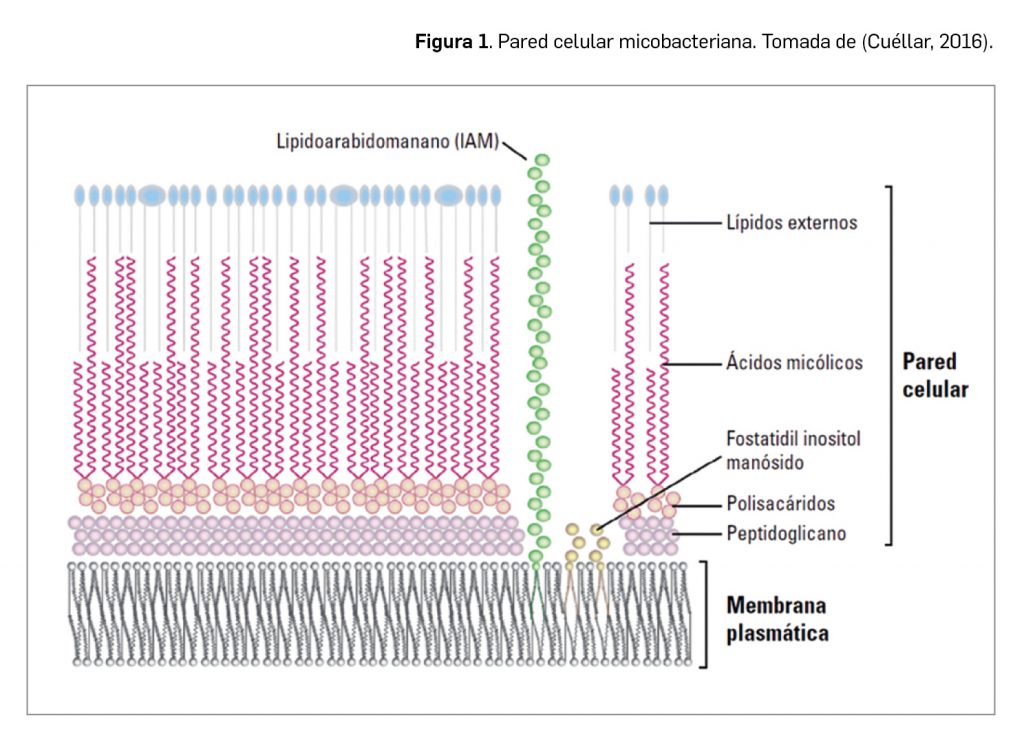
Las especies del complejo M. tuberculosis son patógenos intracelulares y, a nivel estructural, se caracterizan por presentar una pared celular hidrofóbica muy rica en unos lípidos denominados ácidos micólicos, que le confieren propiedades ácido-alcohol resistentes que son relevantes en el proceso de diagnóstico y que también se relacionan con la resistencia a muchos fármacos, desinfectantes, a la desecación y a la congelación.
Tras alcanzar los alveolos pulmonares, el sistema inmunitario del paciente desencadena una respuesta inmunitaria que inicialmente consiste en la fagocitosis por los macrófagos, pero que no suele ser eficaz para la erradicación e incluso puede contribuir a la diseminación de la infección a los ganglios linfáticos regionales, al torrente sanguíneo y a otros órganos y tejidos. La micobacteria presenta un crecimiento lento que estimula una respuesta adaptativa a través de células T y anticuerpos, que salvo en caso de que el foco infeccioso sea muy reducido, tampoco logra erradicar la infección pero sí controlar el crecimiento bacteriano, permaneciendo latente durante largos periodos, de meses o años, hasta que se produce una reducción de la inmunocompetencia.
El único reservorio es el ser humano y el contagio se produce a través de las secreciones respiratorias (tos, estornudos), aunque la capacidad infectiva de este microorganismo es baja, requiriéndose un contacto íntimo y prolongado para que se produzca el contagio.
La tuberculosis es difícil de diagnosticar porque puede simular cualquier otro cuadro infeccioso, con síntomas inespecíficos como fiebre, pérdida de peso o astenia. No obstante, el esputo sanguinolento es un signo característico y también puede aparecer dificultad para respirar y sudoración nocturna. Estos síntomas corresponden a la tuberculosis pulmonar, que es la forma más habitual de presentación. Sin embargo, en ciertos casos, especialmente en individuos inmunodeprimidos, se puede producir tuberculosis extrapulmonar, que puede afectar a cualquier órgano.
La prueba de la tuberculina o de Mantoux permite detectar la infección latente en base a una reacción de hipersensibilidad retardada, pero en la actualidad el principal método diagnóstico para un caso sospechoso es la detección de interferón gamma, dado que un resultado positivo en la prueba de la tuberculina no indica necesariamente infección activa y un resultado negativo tampoco permite descartar una infección reciente. La visualización del bacilo en una muestra de esputo también es útil de cara a establecer el diagnóstico.
La infección por M. tuberculosis se considera actualmente un problema de salud pública a nivel internacional, especialmente por la aparición de casos de multirresistencia (MDR-TB), caracterizados por resistencia a los dos fármacos antituberculosos más utilizados, isoniazida y rifampicina, y de tuberculosis extensamente resistente (XDR-TB), en la que existe resistencia también a los tratamientos inyectables de segunda línea y a las fluoroquinolonas. En España, de acuerdo al Ministerio de Sanidad (MS) en el año 2021 se notificaron 3600 casos de tuberculosis, de los cuales 58 fueron casos de MDR-TB o XDR-TB (MS, 2023). En ocasiones se distingue un escalón intermedio denominado pre-XDR-TB, para casos con tuberculosis resistente a isoniazida y rifampicina y sensible o bien a inyectables de segunda línea o bien a fluroquinolonas, pero no a ambos. Se estima que la mortalidad por tuberculosis no tratada alcanza el 50% y, en el caso de la MDR-TB, a pesar del tratamiento aproximadamente un 15% fallece debido a la enfermedad (OMS, 2022).
El tratamiento de la enfermedad es complejo y requiere de varios meses de duración. Se consideran fármacos antituberculosos de primera línea isoniazida, pirazinamida, rifampicina, rifabutina y estreptomicina durante dos o tres meses, seguidos de cuatro meses de tratamiento de continuación con isoniazida y rifampicina en casos no multirresistentes (Cuéllar, 2016).
La isoniazida tiene un efecto bactericida frente a las micobacterias en crecimiento y bacteriostático durante la fase de reposo. Actúa mediante la inhibición de un precursor de los ácidos micólicos, impidiendo que las células crezcan y se reproduzcan. La pirazinamida es un análogo de la isoniazida que solo presenta actividad bactericida frente a M. tuberculosis intracelular de crecimiento lento y también modifica el pH del medio, lo que dificulta el crecimiento del bacilo. La isoniazida puede provocar reacciones neurotóxicas, como neuritis periférica, por aumento de la excreción de piridoxina, por lo que se administra conjuntamente con vitamina B6. Se utiliza también en la profilaxis.
Las rifamicinas (rifampicina y rifabutina) únicamente presentan actividad bactericida sobre las bacterias en fase de crecimiento. Actúan mediante la inhibición de la síntesis bacteriana de ARNm por interferencia con una ARN polimerasa ADN-dependiente. Esta enzima está también presente en las mitocondrias humanas, pero las rifamicinas no son capaces de atravesar la membrana mitocondrial, lo que les confiere selectividad sobre las micobacterias. La rifampicina produce una coloración rojiza de las secreciones (orina, sudor, saliva, semen, lágrimas) y es un potente inductor enzimático. La resistencia a rifampicina se desarrolla muy rápidamente cuando se utiliza en monoterapia.
El etambutol actúa impidiendo la síntesis de la pared micobacteriana mediante la inhibición competitiva de la transferencia de los ácidos micólicos, por lo que su actividad es selectiva sobre las micobacterias. Únicamente es activo frente a microorganismos en fase reproductiva, no en la fase latente. Una de sus ventajas es que las resistencias a este fármaco se producen más lentamente e incluso es capaz de retrasar la aparición de resistencias a otros antituberculosos, por lo que suele combinarse con isoniazida, rifampicina y/o pirazinamida en el tratamiento de primera línea.
La estreptomicina es un aminoglucósido con efecto tuberculostático sobre los bacilos intracelulares. Las resistencias a este fármaco se producen rápidamente cuando se administra en monoterapia, por lo que siempre se utiliza en asociación, principalmente con isoniazida y/o rifampicina. Su mecanismo de acción es el propio de los aminoglucósidos, esto es, la interferencia con la síntesis proteica al unirse a la subunidad 30S de los ribosomas bacterianos.
La MDR-TB se considera una enfermedad rara en la Unión Europea, Estados Unidos y Japón. En MDR-TB se utilizan fármacos considerados de segunda línea, principalmente los aminoglucósidos inyectables amikacina y kanamicina, las fluoroquinolonas, delamanid y otros fármacos no disponibles en España, como capreomicina, etionamida o cicloserina. Estos fármacos se reservan para casos de resistencia debido a que se consideran en general menos activos frente a las micobacterias y a la elevada toxicidad asociada al tratamiento. El régimen de tratamiento se alarga durante 4-6 meses en una fase intensiva en la que se utiliza un aminoglucósido junto con tres o cuatro fármacos adicionales, incluyendo una fluoroquinolona, seguido de una fase de continuación sin el aminoglucósido inyectable, con una duración total del tratamiento de 18-24 meses (OMS, 2019).
Amikacina y kanamicina comparten mecanismo de acción con estreptomicina, mientras que las fluoroquinolonas actúan sobre la ADN girasa, una topoisomerasa de tipo II, que impide el enrollamiento de las hebras de ADN.
Delamanid es un antituberculoso oral de tipo nitroimidazol que actúa como bactericida al inhibir la síntesis de ácidos micólicos (metoximicólico y cetomicólico).
En resumen, la aparición de resistencias frente a los fármacos habitualmente empleados en esta enfermedad constituye un problema de gran relevancia sanitaria, dada la elevada tasa de mortalidad que la tuberculosis presenta cuando no se establece un tratamiento o en los casos en que este no resulta efectivo, y actualmente se considera que en el tratamiento de la tuberculosis multirresistente existe una necesidad médica no cubierta. Por ello, se requiere de antibióticos que amplíen el arsenal terapéutico frente a esta enfermedad.
Acción y mecanismo
Bedaquilina es un nuevo antibiótico oral con actividad frente a la tuberculosis. Actúa inhibiendo de manera específica a la enzima ATP sintasa micobacteriana, esencial en la producción de energía para el microorganismo, lo que produce un efecto bactericida tanto en las bacterias en reproducción como en las bacterias en reposo (fase latente). Se encuentra actualmente autorizado y está comercializado como medicamento huérfano, estando indicado en pacientes adultos y pediátricos (de 5 a menos de 18 años y un peso de al menos 15 kg) para ser utilizado como parte de un adecuado tratamiento combinado de la tuberculosis pulmonar multirresistente (MDR-TB) cuando un régimen de tratamiento efectivo no puede instaurarse por motivos de resistencia o tolerabilidad.
El fármaco presenta actividad inhibitoria in vitro frente a cepas multirresistentes de M. tuberculosis con una concentración mínima inhibitoria (CMI) de entre ≤ 0,008 y 0,12 μg/ml. Se han observado también in vitro propiedades bactericidas que, a diferencia de lo que ocurre para otros antituberculosos, como isoniazida y rifampicina, parecen más potentes frente a micobacterias latentes que frente a los bacilos en reproducción.
En modelo murino, bedaquilina en monoterapia mostró una eficacia bactericida similar a la combinación de rifampicina, isoniazida y pirazinamida, y superior a la de rifampicina sola. En cobayas, un modelo en el que la fisiopatología de la enfermedad muestra aspectos muy similares a los que produce en humanos, bedaquilina en monoterapia mostró una eficacia incluso superior a la de la combinación mencionada, con una erradicación casi completa de las micobacterias tras seis semanas de tratamiento (EMA, 2013).
Por último, se han descrito como mecanismos de resistencia a bedaquilina mutaciones en el gen atpE, que codifica para la diana de la ATP sintasa, y en el gen Rv0678, que regula la expresión de una bomba de eflujo (AEMPS, 2021).
Aspectos moleculares
Bedaquilina es químicamente el (1R,2S)-1-(6-bromo-2-metoxi-3-quinolil)-4-(dimetilamino)-2-(1-naftalenil)-1-fenil-2-butanol, con la fórmula molecular C32H31BrN2O2 y un peso molecular de 555,50 g/mol.
Se formula junto con ácido fumárico en proporción 1:1. El fumarato de bedaquilina es un polvo no higroscópico blanco o casi blanco. Es prácticamente insoluble en agua y soluble en solventes orgánicos. Se trata de una sustancia de clase 2 de acuerdo al Sistema de Clasificación Biofarmacéutica (con baja solubilidad y alta permeabilidad).
Eficacia y seguridad clínicas
La evaluación de la eficacia y la seguridad clínicas de bedaquilina por vía oral en adultos se ha sustentado en el ensayo C208, de fase 2, controlado por placebo, doble ciego y aleatorizado, y en el estudio C209, de fase 2b y de un solo brazo. Asimismo, se dispone de los datos de seguridad de un ensayo de fase 2 (estudio C211), multicéntrico, de un solo brazo y abierto que incluyó dos cohortes de edad con población pediátrica (entre 5 y 12 años y desde 12 a 18 años).
El estudio C208 incluyó a un total de 207 pacientes con tuberculosis multirresistente por M. tuberculosis, y se dividió en una primera etapa exploratoria (n= 47) en la que el tratamiento con bedaquilina duró 8 semanas y en una segunda etapa (n= 160), considerada como estudio pivotal, en la que el tratamiento duró 24 semanas, iniciado con una dosis intensiva de 400 mg/día durante dos semanas y una posterior de mantenimiento con 200 mg tres veces por semana. El objetivo de la primera etapa fue la evaluación de la farmacocinética, de la actividad antibacteriana, de la seguridad y de la tolerabilidad del fármaco, mientras que en la segunda etapa el objetivo fue la demostración de la superioridad de bedaquilina frente a placebo en un régimen combinado frente a MDR-TB.
Antes del inicio, los pacientes discontinuaron el tratamiento previo con el objetivo de establecer un periodo de lavado de 7 días, y se excluyó a aquellos que habían sido pretratados con fármacos de segunda línea, en tratamiento por arritmias cardiacas y con complicaciones extrapulmonares o neurológicas graves derivadas de la tuberculosis.
En cuanto a las características basales en la población por intención de tratar (ITT), la mayor parte fueron varones (63,1%) y la edad mediana fue de 34 años; hubo un mayor porcentaje de pacientes positivos para la infección por VIH en el grupo de placebo (19,8% vs. 10,1%). En aquellos pacientes en los que se analizaron las características de multirresistencia (n= 111), el 76% presentaba MDR-TB, el 22% presentaba pre-XDR-TB y 3 pacientes se excluyeron de la población ITT por presentar XDR-TB. El tratamiento con bedaquilina se combinó con otros fármacos recomendados en el tratamiento de la MDR-TB, tales como kanamicina, ofloxacino, etionamida, pirazinamida y cicloserina. La variable principal de eficacia fue el tiempo hasta la conversión (negativización) del cultivo de esputo en dos muestras consecutivas; otras variables de eficacia fueron la tasa de conversión del cultivo de esputo, la curación de la enfermedad y el fracaso terapéutico.
Los resultados de la primera etapa del estudio (Diacon et al, 2009) indican una mayor probabilidad de respuesta al tratamiento con bedaquilina frente a placebo, en términos de negativización del cultivo de esputo (HR: 11,77; p= 0,0034) a las 8 semanas de tratamiento. En la segunda fase (Diacon et al., 2014), tras un seguimiento de 24 semanas, el tratamiento con bedaquilina resultó en una reducción estadísticamente significativa (p< 0,0001) del tiempo mediano hasta la conversión del cultivo de esputo respecto a placebo (83 días vs. 125 días). Respecto a los objetivos secundarios, la tasa de conversión a las 24 semanas fue también superior al añadir bedaquilina (79% vs. 58%; p= 0,008), con una diferencia –estadísticamente significativa– sostenida incluso 6 meses tras el final del tratamiento (62% vs. 44%; p= 0,035). Las tasas de conversión en la semana 24 fueron más altas en un régimen combinado con otros tres fármacos antituberculosos (tasa de respuesta: 85% vs. 64% con placebo) o cuatro (88% vs. 31%); con un quinto fármaco adicional no se observó una mejora en la tasa de respuesta, que incluso fue superior en el brazo de placebo (75% vs. 80%).
Por otro lado, el estudio C209 (Pym et al., 2016) fue un ensayo abierto de fase 2b, de un solo brazo (N= 233), con la misma pauta posológica que en la etapa 2 del estudio C208 y una duración de entre 72 y 96 semanas (al menos, 12 meses tras la negativización del cultivo de esputo). La variable principal de eficacia fue el tiempo hasta la conversión del cultivo de esputo a las 24 semanas de tratamiento. Con datos de una muestra de 205 pacientes (población por intención de tratar modificada), el tiempo mediano hasta la conversión fue de 57 días, verificándose una tasa de respuesta del 75%. Los datos de conversión en función del número de agentes antituberculosos utilizados son similares a los del estudio C208.
Se dispone también de los datos más recientes de un estudio de fase 3 aleatorizado, ciego y de no inferioridad (Goodall et al., 2022) en el que se analizó la eficacia del tratamiento para MDR-TB con bedaquilina en comparación con un régimen de control sin bedaquilina durante 9 meses en 588 pacientes. Los resultados de eficacia, determinada como la tasa de cultivos negativos en la semana 76 tras la aleatorización, mostraron superioridad de los dos regímenes que incluyeron bedaquilina, con un 83% de respuestas positivas en el régimen de bedaquilina (9 meses) y un 71% en el brazo de control (p< 0,0001).
Adicionalmente, los resultados del estudio C211 han permitido extender la indicación de uso a la población pediátrica de entre 5 y 18 años de edad. En niños y adolescentes de esos grupos etarios, la Agencia Europea de Medicamentos (EMA, 2020; EMA, 2021) consideró apropiado extrapolar los resultados de eficacia obtenidos en adultos con una dosis que permita obtener el mismo nivel de exposición.
Por otra parte, los datos de seguridad del nuevo fármaco en adultos provienen de un total de 380 pacientes con tuberculosis multirresistente tratados en estudios de fase 2 y de 265 participantes sanos que recibieron una dosis única o múltiple. En los estudios C208 y C209, la proporción de pacientes que sufrió algún evento adverso fue muy alta (>95%). En concreto, en el estudio C208 se observó una mayor proporción de determinados eventos adversos en el brazo de bedaquilina respecto al de placebo, principalmente artralgias (33% vs. 22%), dolor de cabeza (28% vs. 12%) y hemoptisis (18% vs. 11%); los eventos adversos graves fueron también más comunes en el brazo de bedaquilina (7% vs. 2% con placebo), aunque no se identificó ningún evento adverso que considerado individualmente tuviera una frecuencia superior al 1%. En la segunda etapa del estudio C208 se reportaron 10 muertes entre pacientes tratados con bedaquilina (vs. 2 en el brazo de placebo), pero ninguna de ellas se relacionó con el nuevo fármaco, pudiendo tratarse de un efecto del reducido tamaño muestral.
En los dos estudios con población adulta, el tratamiento con bedaquilina se asoció a un incremento significativo, aunque asintomático, del intervalo QTc1 (diferencia mediana de +10 ms frente a placebo a la semana 18), aspecto que debe ser tenido en cuenta en su manejo. Por otro lado, se ha identificado un incremento de las transaminasas hepáticas con el uso de bedaquilina en comparación con placebo, y 3 pacientes en el estudio C208 (de un total de 102) abandonaron el tratamiento debido a efectos adversos hepáticos.
En la población pediátrica, se dispone de los datos de 30 pacientes (15 en la cohorte de entre 5 y 12 años y 15 en la cohorte de 12-18 años), de modo que el reducido tamaño muestral limita la extracción de conclusiones respecto a la frecuencia de los eventos adversos observados. En cualquier caso, la frecuencia de los mismos fue similar a la vista en los estudios en adultos, y no parece que el perfil de seguridad difiera sustancialmente para un mismo nivel de exposición sistémica. En niños no se vieron cambios significativos en el electrocardiograma durante el uso del fármaco.
Aspectos innovadores
Bedaquilina es un nuevo antibiótico oral activo frente a la infección por M. tuberculosis multirresistente. Su mecanismo de acción consiste en la inhibición específica de la ATP sintasa micobacteriana, esencial en la producción de energía para el microorganismo, lo que produce un efecto bactericida tanto en las bacterias en reproducción como en las bacterias en reposo (fase latente). Se encuentra actualmente autorizado y está comercializado como medicamento huérfano, estando indicado en pacientes adultos y pediátricos (desde 5 años hasta menos de 18 años y un peso de al menos 15 kg) para ser utilizado como parte de un adecuado tratamiento combinado de la tuberculosis pulmonar multirresistente (MDR-TB) cuando un régimen de tratamiento efectivo no puede instaurarse por motivos de resistencia o tolerabilidad.
Su autorización se sustentó en los datos clínicos de un estudio de fase 2, que analizó la eficacia de bedaquilina frente a placebo en un régimen multifármaco en pacientes con MDR-TB y pre-XDR-TB en términos de la reducción del tiempo hasta la conversión del cultivo de esputo. En la segunda etapa del estudio (fase 2b, considerada pivotal), el tratamiento con bedaquilina redujo de manera estadísticamente significativa (p< 0,0001) la mediana de tiempo hasta la obtención de un cultivo negativo respecto a placebo, en más de 40 días (83 vs. 125 días). La tasa de conversión fue también significativamente superior al añadir bedaquilina (79% vs. 58%; p= 0,008), con una diferencia sostenida en torno a 20 puntos porcentuales incluso 6 meses tras el final del tratamiento (62% vs. 44%; p= 0,035). Otro estudio, también de fase 2b y de un solo brazo, reveló que la mediana del tiempo hasta la conversión del cultivo con un tratamiento con bedaquilina fue de 57 días: esta mayor reducción respecto al estudio pivotal podría explicarse por el hecho de que los pacientes recibieron un tratamiento más individualizado, lo que se asemeja en mayor medida a la práctica clínica habitual (AEMPS, 2023).
Los datos de eficacia en función del número de fármacos usados de forma concomitante apuntan a que los resultados son óptimos en un régimen combinado con tres antituberculosos adicionales, incluyendo a ser posible un aminoglucósido inyectable (amikacina o kanamicina) y/o una fluoroquinolona, considerados más activos frente a las formas multirresistentes de la tuberculosis que otros fármacos de segunda línea. En pacientes en los que no es posible utilizar ninguno de esos fármacos por motivo de intolerancia o de resistencia la adición de bedaquilina a un régimen combinado de tratamiento podría ser la única opción de llegar a curar la infección multirresistente (EMA, 2013).
Como para el resto de antibacterianos, la aparición de resistencias frente a bedaquilina puede comprometer su potencial eficacia. Los datos clínicos, aunque todavía prematuros, apuntarían a una baja frecuencia de aparición de resistencias con este nuevo fármaco. Es más, parece que hasta cierto punto el tratamiento con bedaquilina puede retrasar la aparición de resistencias frente a otros fármacos; de hecho, en la segunda etapa del estudio pivotal se observó la aparición de resistencia a fármacos distintos a bedaquilina en 2 casos de 10 pacientes en el brazo de bedaquilina vs. 16 casos de 31 pacientes en el brazo de placebo.
Los estudios que han permitido obtener la indicación pediátrica se encuentran todavía en marcha y en ellos se ha considerado que la eficacia y seguridad del fármaco son extrapolables a partir de los resultados obtenidos en población adulta, con el debido ajuste de dosis que garantice una exposición sistémica similar.
En cuanto al perfil toxicológico del nuevo fármaco, durante los estudios clínicos de fase 2 se observó una elevada proporción de eventos adversos, cercana al 100%, aunque similar en el brazo de tratamiento y en el de placebo. Las reacciones adversas asociadas con mayor frecuencia al fármaco fueron artralgias, dolor de cabeza y hemoptisis. Los eventos graves, poco frecuentes en general, ocurrieron en mayor medida en el brazo de bedaquilina (7% vs. 2%), pero ninguno de ellos con una frecuencia superior al 1%. La seguridad de bedaquilina parece similar en la población pediátrica, aunque no se puede concluir al respecto por las limitaciones de los estudios llevados a cabo en este grupo de la población (pequeño tamaño muestral).
Dentro del perfil de seguridad del fármaco preocupa especialmente su potencial arritmogénico, derivado de su capacidad para alargar el intervalo QT, así como una potencial hepatotoxicidad, aunque se reportó una baja frecuencia de eventos adversos asociados a estos hallazgos clínicos. La mayor proporción de fallecimientos notificada respecto a placebo en el estudio pivotal, aunque sin relación de causalidad confirmada, también hace necesario disponer de datos adicionales de estudios de fase 3 que caractericen mejor el balance beneficio-riesgo del fármaco. Por este motivo, la indicación se ha restringido a aquellos pacientes en los que no se pueda establecer un régimen alternativo para MDR-TB por motivos de tolerabilidad o resistencia (EMA, 2013).
En resumen, bedaquilina es la primera arilquinolina aprobada para el tratamiento de la tuberculosis multirresistente, con un innovador mecanismo de acción en su indicación que se basa en la inhibición de la ATP sintasa de M. tuberculosis. Su inclusión en un régimen combinado de tratamiento, especialmente junto con un aminoglucósido inyectable y/o una fluoroquinolona, permitirá incrementar la eficacia en la erradicación de las micobacterias. Además, cuenta con un perfil de seguridad que, aun con incertidumbres respecto al potencial arritmogénico y hepatotoxicidad, parece aceptable en relación al beneficio clínico aportado. En el contexto de un tratamiento complejo y de larga duración en el que la aparición de resistencias es frecuente, bedaquilina puede contribuir a afrontar una necesidad médica no cubierta. No obstante, con una indicación por ahora limitada a la ausencia de alternativas, no parece que vaya a suponer un cambio disruptivo en la práctica clínica diaria frente a la tuberculosis multirresistente.
Valoración