Resumen
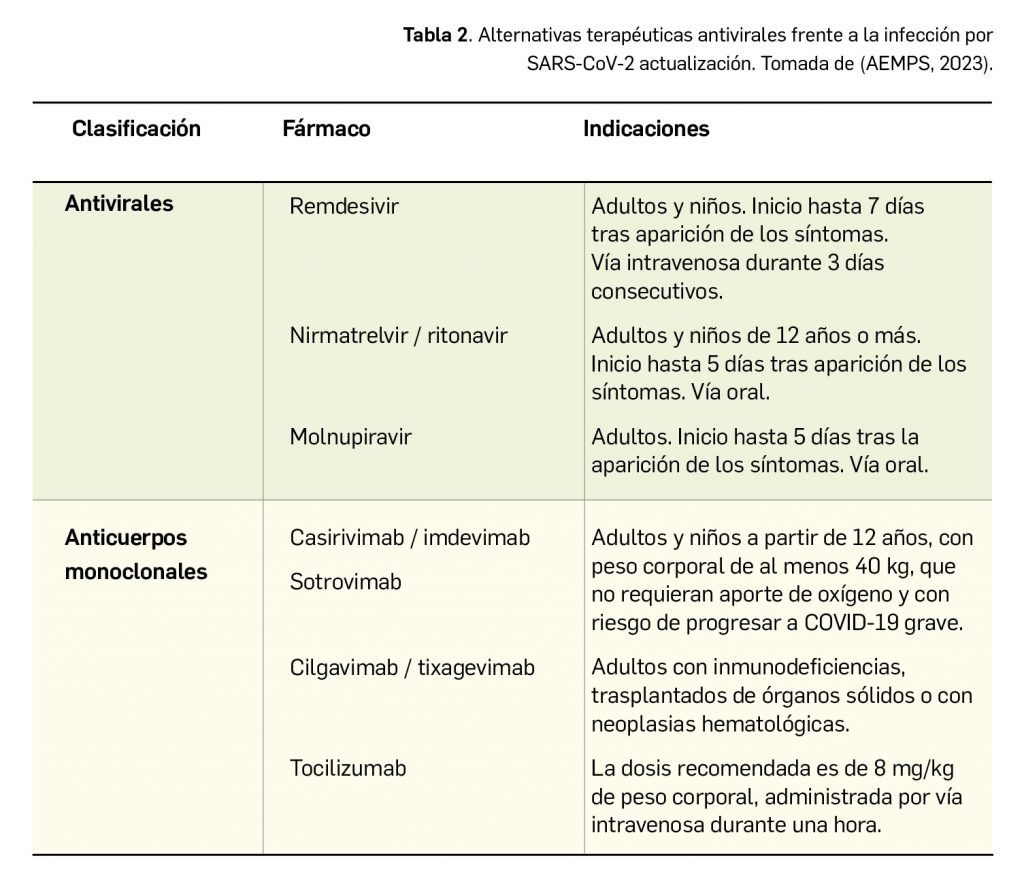
Cefiderocol es una nueva cefalosporina con actividad antibiótica frente a bacterias gramnegativas. Presenta una vía original de entrada al interior celular mediante transporte activo como consecuencia de las características sideróforas de su cadena lateral, capaz de captar el hierro libre extracelular, que complementa a la habitual difusión pasiva a través de las porinas de la membrana externa del microorganismo de las cefalosporinas. En todo caso, su mecanismo de acción farmacológica es el propio de los fármacos de ese grupo –de todos los antibióticos betalactámicos–, o sea, la inhibición de la síntesis de la pared celular por unión a las proteínas de fijación de las penicilinas (PFP). En base a estas acciones, cefiderocol ha sido autorizado con indicación en el tratamiento por vía intravenosa y a nivel intrahospitalario de infecciones debidas a microorganismos gramnegativos aerobios en adultos con opciones terapéuticas limitadas, siguiendo siempre las guías oficiales relativas al uso correcto de antibacterianos.
La eficacia y la seguridad clínicas de cefiderocol se han evaluado en dos estudios de fase 3 y en uno de fase 2. En una comparación con imipenem/cilastatina (ensayo de fase 2) en infecciones complicadas del tracto urinario o pielonefritis no complicadas se determinó la no inferioridad de cefiderocol respecto a la variable primaria de eficacia, que fue la proporción de respuesta clínica y microbiológica en el test de curación, al observarse mayor proporción de respuestas en el brazo de cefiderocol (72,6% vs. 54,6%). También se comparó el nuevo antibiótico con la mejor alternativa disponible en diversos tipos de infección (neumonía, sepsis, infección urinaria complicada), con datos similares de eficacia en ambos brazos, pero mayor mortalidad en el brazo de cefiderocol. Por otro lado, en una comparación con meropenem, cefiderocol resultó no inferior en cuanto respecta a la mortalidad por cualquier causa en pacientes con neumonía por bacterias gramnegativas.
El perfil de seguridad se considera aceptable, siendo los eventos adversos observados en los ensayos clínicos similares a los de los comparadores activos. Las reacciones asociadas al tratamiento no parecen diferir sustancialmente de las de otras cefalosporinas (diarrea, rash cutáneo, reacciones de hipersensibilidad, disminución del umbral convulsivo y elevación de transaminasas); mayoritariamente fueron de intensidad leve o moderada y salvo en muy raras ocasiones no motivaron la discontinuación del tratamiento.
Los resultados de los ensayos clínicos presentan importantes limitaciones debido al tipo de diseño y a los tamaños muestrales y la EMA consideró como decisivos los datos de los ensayos preclínicos a la hora de emitir una opinión positiva. A pesar de que cefiderocol plantea una novedosa forma de entrada a las bacterias gramnegativas que podría permitir solventar algunos de los mecanismos habituales de resistencia en estos microorganismos, no es descartable que en algunos casos se pueda producir resistencia de manera rápida, incluso intra-tratamiento. La utilidad de cefiderocol podría ser superior en patógenos productores de metalo-betalactamasas, para los que las alternativas terapéuticas son muy limitadas.
Por tanto, aunque se requiere de resultados más exhaustivos procedentes de la práctica clínica habitual, no parece que cefiderocol vaya a constituir una alternativa interesante en infecciones producidas por bacterias gramnegativas multirresistentes, más allá de los casos en que otros tratamientos para los que se disponga de mayor experiencia hayan fracasado (por resistencias) o estén desaconsejados (por intolerancia).
Aspectos fisiopatológicos
En los últimos años está adquiriendo una relevancia sanitaria y social cada vez mayor la resistencia microbiana a los antibióticos, por constituir una grave amenaza para la salud pública a nivel global –y a nivel regional europeo– con consecuencias como un aumento de los costes sanitarios, motivado sobre todo por una hospitalización más larga de los pacientes y, lo más preocupante, un aumento de la mortalidad por infecciones.
En este sentido, los Centros para la Prevención y Control de Enfermedades de EE.UU. estiman que en aquel país se producen cada año más de 2 millones de infecciones por bacterias multirresistentes, que son causa de más de 23 000 muertes; en la Unión Europa, el ECDC (European Centre for Disease Prevention and Control) calcula que en 2015 se produjeron más de 700 000 infecciones y 33 000 muertes por la multirresistencia de los microorganismos a los antibióticos disponibles. Y en España, los datos recopilados por la Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica (SEIMC) permiten estimar que en 2018 fallecieron en España aproximadamente 35 000 personas por esta causa, sobre un total de 180 600 infecciones causadas por bacterias multirresistentes.
El pronóstico de futuro no es nada halagüeño. Las resistencias a los agentes antimicrobianos han ido en progresivo aumento en las últimas décadas y preocupa especialmente el caso de las bacterias gramnegativas, debido a la frecuencia con la que algunos de estos patógenos generan resistencia a la mayor parte o incluso a todos los tratamientos disponibles, por múltiples mecanismos. Entre estas bacterias multirresistentes destacan algunas como Pseudomonas aeruginosa1, Klebsiella pneumoniae2, Acinetobacter baumannii y las de la familia Enterobacteriaceae (enterobacterias), incluida Escherichia coli.
Las infecciones causadas por bacilos gramnegativos multirresistentes pueden ser muy variadas, principalmente en el ámbito hospitalario o relacionadas con la asistencia sanitaria. En las que más habitualmente se ven implicados son: infecciones del tracto urinario complicadas (ITUc), infecciones respiratorias severas, especialmente neumonías nosocomiales, infecciones intraabdominales o septicemias (originadas por infecciones de localización quirúrgica o de dispositivos intravasculares como catéteres).
La resistencia al tratamiento antibiótico puede clasificarse en función del tipo de mecanismo como enzimática o no enzimática. Las resistencias basadas en mecanismos enzimáticos suelen consistir en la expresión de proteínas que inactivan al fármaco (como las betalactamasas) o de bombas de eflujo que expulsan al fármaco hacia el exterior de la célula bacteriana, impidiendo su acción. Esta expresión puede ser consecuencia de mutaciones cromosómicas o de la transferencia de genes de resistencia, frecuentemente a través de plásmidos.
Desde el punto de vista del tratamiento, los antibióticos betalactámicos han sido tradicionalmente uno de los grupos de antibióticos más empleados frente a infecciones provocadas por patógenos gramnegativos. La producción de betalactamasas es el principal mecanismo de resistencia frente a los antibióticos betalactámicos, incluyendo la expresión de genes como AmpC y betalactamasas de espectro extendido (BLEEs), capaces de inactivar a todos los antibióticos de este grupo, con excepción de los carbapenemes, que se considerarían de elección frente a infecciones por enterobacterias productoras de BLEEs, aunque hay otras opciones, sobre todo frente a ITU (por ejemplo, aminoglucósidos y piperacilina/tazobactam).
Sin embargo, el aumento de uso de los antibióticos carbapenémicos –considerados como la familia de antibióticos con más amplio espectro antibacteriano frente a gramnegativas– se ha acompañado a su vez de un aumento en la producción de carbapenemasas en esas bacterias, que ha limitado todavía más las alternativas de tratamiento. Las combinaciones de antibióticos betalactámicos con inhibidores de las betalactamasas de introducción más reciente, como ceftolozano/tazobactam, ceftazidima/avibactam o meropenem/vaborbactam pueden resultar útiles en algunos casos de bacterias productoras de cabapenemasas, siempre considerando los distintos perfiles de cobertura frente a los distintos tipos de carbapenemasas, pero presentan una eficacia muy limitada frente a algunos tipos de bacterias productoras de metalo-betalactamasas. El recurso a otros grupos de antibióticos, como las fluorquinolonas, las tetraciclinas3 o la fosfomicina4, es también una opción cada vez más limitada, lo que finalmente lleva a optar por tratamientos asociados a mayor toxicidad, como los aminoglucósidos (por ejemplo, amikacina) o las polimixinas (colistina5), que además de no ser aptas para todos los pacientes, también han visto reducida su eficacia en numerosas cepas bacterianas que han acabado por desarrollar mecanismos de resistencia (AEMPS, 2023).
En un contexto de aumento de la mortalidad como consecuencia del fracaso farmacoterapéutico en infecciones causadas por bacterias gramnegativas se entiende, por tanto, la importancia de desarrollar nuevos antibióticos capaces de superar los múltiples mecanismos de resistencia de este tipo de microorganismos.
Acción y mecanismo
Cefiderocol es un nuevo antibiótico del grupo de las cefalosporinas que presenta actividad siderófora: se une al hierro libre extracelular a través de su cadena lateral, lo cual le permite penetrar en las bacterias gramnegativas usando los sistemas de transporte activo al espacio periplásmico para la captación de sideróforos, además de poder acceder mediante la vía común de acceso de los antibióticos a microorganismos gramnegativos (difusión pasiva a través de las porinas presentes en la membrana externa). Una vez en el interior celular, como todos los antibióticos betalactámicos, actúa inhibiendo la síntesis de la pared celular bacteriana mediante su unión a las proteínas de fijación de las penicilinas (PFP), produciendo la lisis y la muerte celular. En base a estas acciones, cefiderocol ha sido autorizado con indicación en el tratamiento de infecciones debidas a microorganismos gramnegativos aerobios en adultos con opciones terapéuticas limitadas, siguiendo siempre las guías oficiales relativas al uso correcto de antibacterianos.
Conviene recordar, en este punto, que la cubierta de las bacterias varía sustancialmente de una especie a otra, pero colectivamente se aprecian diferencias cualitativas entre bacterias grampositivas y gramnegativas. Estas últimas disponen de una membrana citoplásmica similar a la de las células eucariotas: rodeando a la membrana celular existe lo que se conoce como espacio periplásmico, que a su vez está encerrado por una red de peptidoglucano y, finalmente, por una membrana exterior. En cambio, la cubierta de las grampositivas es más simple, pero contiene una capa mucho más tupida de peptidoglucano, formada por fibras poliméricas estrechamente entrelazadas que forman una estructura reticular cuya misión esencial es proporcionar rigidez a la cubierta bacteriana, y permite a la bacteria sobrevivir en entornos químicos agresivos. Dado que la osmolaridad citoplasmática bacteriana es de casi 3 veces la de las células eucariotas, sin la existencia de una pared celular y su red de peptidoglucano, el protoplasto bacteriano se hincharía y estallaría, debido a su hipertonicidad con respecto al medio (generalmente hipotónico), conduciendo a la muerte de la célula bacteriana. Se comprende, por tanto, que el efecto inhibitorio de cefiderocol sobre la síntesis correcta de la pared bacteriana subyace tras su efecto bactericida.
En estudios in vitro en cepas de P. aeruginosa se ha observado que cefiderocol, debido a su forma de entrada a la célula, fue capaz de superar determinados mecanismos de resistencia habituales en bacterias gramnegativas, como la regulación a la baja del número de porinas o la expresión de bombas de eflujo. En estos estudios también se ha observado que cefiderocol no se ve afectado por la mayor parte de las betalactamasas, incluyendo las metaloenzimas (AEMPS, 2022).
A priori, el fármaco presenta actividad intrínseca frente a bacilos gramnegativos, tanto fermentadores (Enterobacterales) como no fermentadores (P. aeruginosa, A. baumannii y S. maltophilia), habiéndose descrito valores de puntos de corte de sensibilidad inferiores a 2 y a 4 mg/l. Estudios sobre la sensibilidad de distintas cepas de gramnegativos han mostrado que los porcentajes de susceptibilidad al tratamiento con cefiderocol son cercanos al 100%, incluso en bacterias habitualmente resistentes a las alternativas disponibles en la antibioterapia frente a infecciones por gramnegativos, como ceftazidima/avibactam, ceftolozano/tazobactam, ciprofloxacino o colistina.
Estudios in vivo en modelos animales han probado reducciones de la carga bacteriana dependientes de la dosis y un aumento de la eficacia en infusiones prolongadas, lo que parece sugerir un perfil farmacocinético y farmacodinámico tiempo-dependiente para este antibiótico (EMA, 2020). Se acepta que las resistencias actuales a cefiderocol son bajas (< 10%), aunque variables dependiendo del estudio y cepas estudiadas. Los mecanismos de resistencia al fármaco se pueden mediar por la presencia en las bacterias de proteínas PFP mutantes o adquiridas, betalactamasas capaces de hidrolizar cefiderocol, mutaciones que afectan a la captación de hierro o mutaciones en las proteínas de transporte de sideróforos o sobreexpresión de sideróforos bacterianos nativos. La actividad de cefiderocol frente a bacterias grampositivas o anaerobias es pequeña o nula debido a la resistencia intrínseca.de resistencia al fármaco se pueden mediar por la presencia en las bacterias de proteínas PFP mutantes o adquiridas, betalactamasas capaces de hidrolizar cefiderocol, mutaciones que afectan a la captación de hierro o mutaciones en las proteínas de transporte de sideróforos o sobreexpresión de sideróforos bacterianos nativos. La actividad de cefiderocol frente a bacterias grampositivas o anaerobias es pequeña o nula debido a la resistencia intrínseca.
Aspectos moleculares
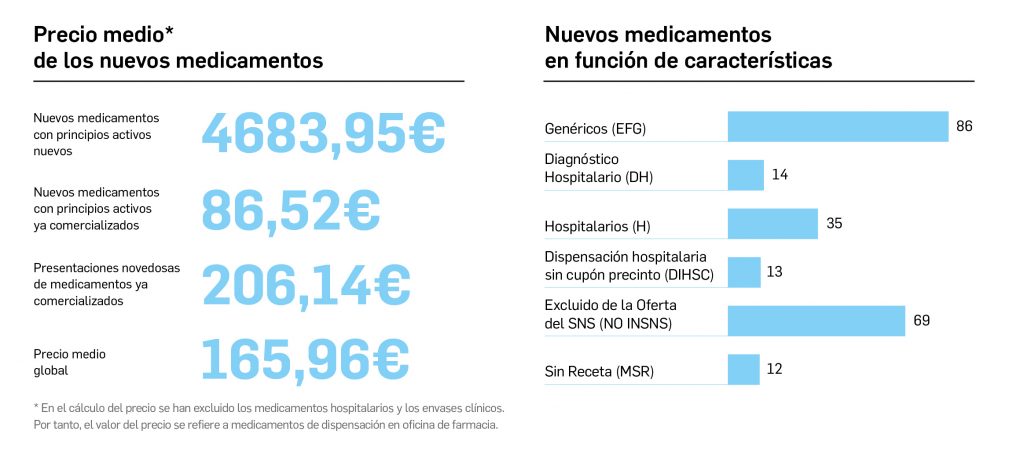
Desde el punto de vista de su estructura química, cefiderocol, que se formula en forma de sulfato tosilato, es el tris[6R,7R)-7-2Z)-2-(2-amino-1,3-tiazol-4-il)-2-{[(2-carboxipropan-2-il)oxi]imino}acetamido]-3-({1-[-cloro-3,4-dihidroxibenzamido)etil]pirrolidin-1-io-1-il}metil)-8-oxo-5-tia-1-azabiciclo[4.2.0]oct-2-eno-2-carboxilato] tetrakis(4-metilbencenosulfonato) hidrato monosulfato, y se corresponde con la fórmula molecular 3·C30H34ClN7O10S2·4C7H8O3S·H2SO4·xH2O. Su masa molecular relativa es de 3043,50 g/mol (anhidro) y su estructura, que incorpora un grupo catecol, puede observarse de forma comparada con otras cefalosporinas en la Figura 1.
Cefiderocol en forma pura se presenta como un polvo cristalino, higroscópico, sensible a la luz y a la hidrólisis y ligeramente soluble en agua. Presenta dos centros quirales y se aísla como un único enantiómero (R,R).
El grupo catecol en posición 3 se considera esencial para la actividad diferencial del antibiótico respecto a otras cefalosporinas, pues es el responsable de la captación de hierro por quelación que permite el acceso al interior de las células bacterianas gramnegativas por transporte activo. También se considera que el grupo catecol está relacionado con la estabilidad del fármaco frente a metalo- y serin-betalactamasas.
La estructura de tipo zwitteriónico de cefiderocol, como ocurre con otras cefalosporinas como ceftolozano, cefepima o ceftazidima, le permite también atravesar con facilidad y de forma rápida los canales porínicos de las bacterias gramnegativas.
Eficacia y seguridad clínicas
La eficacia y la seguridad clínicas de cefiderocol por vía intravenosa en su pauta autorizada han sido evaluadas en tres ensayos pivotales independientes entre sí.
El estudio APEKS-cUTI (Portsmouth et al., 2018) fue un ensayo de fase 2 de no inferioridad, controlado, aleatorizado, doble ciego, multinacional y multicéntrico en el que se comparó el tratamiento con cefiderocol frente a la asociación imipenem/cilastatina (IMP-CS) en pacientes adultos hospitalizados con diagnóstico clínico de infección del tracto urinario complicada (ITUc) o pielonefritis aguda no complicada causada por patógenos gramnegativos que requerían inicio de antibioterapia intravenosa. Aleatorizó a un total de 452 pacientes adultos en proporción 2:1 a recibir cefiderocol o el comparador activo; 371 de ellos se incluyeron en el análisis por de la población ITTm (por intención de tratar modificada), 252 tratados con el fármaco y 119 en el brazo de IMP-CS. Aunque los microorganismos más frecuentemente aislados fueron Escherichia coli (60-66%) y K. pneumoniae (20%), se excluyó del estudio a pacientes con microorganismos resistentes a carbapenemes; también se excluyeron pacientes con más de 2 uropatógenos en el urocultivo, presencia de infección fúngica o insuficiencia renal grave (aclaramiento de creatinina < 20 ml/min).
Las características basales de los pacientes estuvieron correctamente equilibradas entre ambos grupos de tratamiento, con aproximadamente la mitad de los pacientes entre 65 y 75 años y diagnóstico de ITUc sin pielonefritis. E. coli fue el microorganismo más aislado (en casi dos de cada tres pacientes), seguido de K. pneumoniae (≈20%).
En el análisis de eficacia, la variable principal se definió como la respuesta clínica6 y microbiológica –variable compuesta– en el test de curación en el día 7 (±2 días) tras el final del tratamiento, fijándose márgenes del 15% y del 20% en la diferencia respecto al control como criterios de no inferioridad. Entre las variables secundarias se midieron la respuesta clínica y microbiológica precoz, al final del tratamiento y durante el seguimiento (en el día 14 tras el fin del tratamiento), y también la seguridad.
Los resultados evidenciaron que, con una duración media del tratamiento de 9 días (±3) en ambos grupos, se produjo una proporción de respuestas significativamente mayor en el brazo de cefiderocol (72,6% vs. 54,6%), por lo que se cumplió el criterio de no inferioridad. La respuesta clínica fue similar en ambos grupos (90% vs. 87%). En términos de erradicación microbiológicas, a pesar de que se obtuvieron porcentajes superiores al 90% durante el tratamiento (día 4) y al final del tratamiento, en la visita de seguimiento esos porcentajes descendieron notablemente, aunque fueron más elevados en el brazo de cefiderocol (57% vs. 44%).
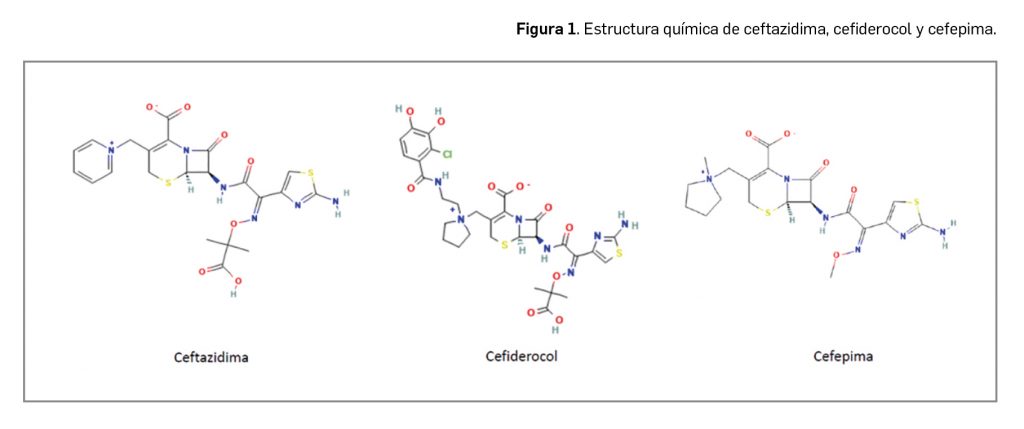
Por otra parte, el estudio pivotal de fase 3 CREDIBLE-CR (Bassetti et al., 2021), de fase 3, aleatorizado, abierto, de grupos paralelos, multinacional y multicéntrico, comparó cefiderocol con la mejor alternativa disponible (MAD) en pacientes adultos con infecciones graves provocadas por bacterias gramnegativas resistentes a carbapenemes. Tuvo un carácter descriptivo.
En el estudio se aleatorizó a 152 pacientes adultos, de los cuales 101 recibieron cefiderocol7 y 51 la MAD; sin embargo, se produjeron varias exclusiones por incumplimiento del protocolo que redujeron el brazo de cefiderocol a 80 pacientes y el de la MAD a 38 pacientes. De cara al análisis de la eficacia se consideraron tres subgrupos, dependiendo de la enfermedad (neumonía, bien fuer intrahospitalario o asociada a ventilación mecánica o cuidados sanitarios, ITUc o sepsis provocada por infección distinta a ITUc). Se excluyeron pacientes que hubieran recibido antibióticos potencialmente activos frente a gramnegativos resistentes a carbapenemes desde 72 h antes del inicio.
Las variables principales de eficacia fueron la proporción de pacientes con neumonía, bacteriemia o sepsis que alcanzó la curación clínica en el día 7 (±2 días) tras el final del tratamiento, y la erradicación microbiológica en pacientes con ITUc (definida como reducción de uropatógenos desde 105 hasta < 103 UFC/ml) también en el día 7 (±2 días) tras finalizar el tratamiento. Como variable secundaria, se evaluó la mortalidad en los días 14 y 28 tras el diagnóstico. Los patógenos gramnegativos más frecuentemente aislados fueron A. baumannii, K. pneumoniae y P. aeruginosa.
Los resultados pusieron de manifiesto porcentajes de curación en los pacientes con neumonía y sepsis que fueron similares en ambos brazos de tratamiento, mientras que la erradicación microbiológica en pacientes con ITUc fue superior en el brazo de cefiderocol (Tabla 1). Un hallazgo bastante revelador, que cuestiona la eficacia del nuevo fármaco, fue que la mortalidad (variable secundaria) fue superior en el brazo de cefiderocol en la visita final del estudio (33% vs. 18%), con una marcada diferencia en los pacientes con infección por A. baumannii (50% vs. 18%).
Finalmente, se efectuó otro estudio pivotal de fase 3 (APEKS-NP), con diseño multicéntrico, aleatorizado, de no inferioridad y doble ciego en el que se comparó la mortalidad por cualquier causa y se evaluaron los resultados clínicos y microbiológicos del tratamiento con cefiderocol o con meropenem como control activo en pacientes adultos ingresados en el hospital con neumonía bacteriana aguda. La población ITTm comprendió a un total de 292 pacientes, asignados al azar (1:1) a cada uno de los brazos de tratamiento, excluyéndose los casos de neumonía extrahospitalaria, atípica o viral y los pacientes con neumonías causadas por bacterias gramnegativas resistentes a carbapenemes en el momento de la aleatorización. Los microorganismos aislados más frecuentemente fueron K. pneumoniae, P. aeruginosa, y A. baumanii.
La variable principal de eficacia fue la mortalidad por cualquier causa en el día 14 tras el inicio del tratamiento. Se analizaron como variables secundarias los resultados clínicos y microbiológicos precoces (en los días 3 o 4 de tratamiento), al finalizar el tratamiento y al final del seguimiento en el día 14 tras el fin del tratamiento.
Con un duración media del tratamiento antibiótico de 10 días (±4), los resultados de mortalidad por cualquier causa en el día 14 fueron similares en ambos brazos de tratamiento: un 12,4% de eventos en los pacientes tratados con cefiderocol y un 11,6% en los tratados con meropenem; se obtuvo un resultado estadísticamente significativo en el análisis de no inferioridad de cefiderocol (p= o,002; dentro del margen predefinido del 12,5% de diferencia), pero no en el de superioridad (p= 0,832). La mortalidad a los 28 días fue también similar, muy ligeramente superior en el brazo de cefiderocol (21,0% vs. 20,5%), sin diferencias reseñables de eficacia en los distintos subgrupos de pacientes considerados (por ejemplo, estancia en UCI o antibioterapia empírica previa, entre otros).
Con respecto a las variables secundarias, en el momento de finalizar el tratamiento, se consideró curada a una mayor proporción de pacientes en el brazo de meropenem (81,0% vs. 77,2%). No obstante, durante el periodo de seguimiento el estado de curación se confirmó en ambos brazos con una frecuencia similar (58%), con un elevado porcentaje de pacientes en los que no se podía determinar claramente el éxito o fracaso clínico (20%) (Wunderink et al., 2021).
En cuanto al análisis del perfil de seguridad, se dispuso de datos de un total de 549 pacientes tratados con cefiderocol. Los eventos adversos observados en los estudios clínicos se produjeron con frecuencias similares en los distintos brazos de tratamiento, sin grandes diferencias en la comparativa entre cefiderocol y los antibióticos usados como comparadores activos. Los eventos adversos relacionados con el nuevo fármaco son consistentes con el perfil de seguridad conocido para las cefalosporinas. Aunque en líneas generales la incidencia fue baja, destacan como reacciones adversas notificadas con mayor frecuencia las siguientes: diarrea, vómitos, reacciones de hipersensibilidad, rash cutáneo, convulsiones y elevación de transaminasas.
La incidencia de eventos adversos graves difirió entre los distintos estudios, siendo relativamente baja en el estudio APEKS-cUTI (5% en el brazo de cefiderocol vs. 8% en el brazo de IMP-CS), pero más alta en los estudios CREDIBLE-CR (50% en los tratados con cefiderocol vs. 47% en los tratados con la MAD) y APEKS-NP (37% con cefiderocol vs. 30% con meropenem). No obstante, las interrupciones del tratamiento debidas a eventos adversos fueron bajas (2% en ambos brazos en el estudio CREDIBLE-CR).
Uno de los aspectos que más preocupa en cuanto respecta a la seguridad de cefiderocol es la elevada mortalidad en comparación con el grupo de control, observada especialmente en el estudio CREDIBLE-CR, en el cual un total de 43 pacientes experimentó eventos adversos que se iniciaron antes del final del tratamiento y 34 con resultado de fallecimiento (34%) en el brazo de cefiderocol y 9 (18%) en el brazo de la mejor terapia disponible, con una diferencia especialmente marcada cuando el agente patógeno fue A. baumannii. Sin embargo, no parece haber resultados respecto a la seguridad de cefiderocol que expliquen esta diferencia.
Aspectos innovadores
Cefiderocol es una nueva cefalosporina con actividad antibiótica frente a bacterias gramnegativas. Presenta una vía original de entrada al interior celular mediante transporte activo como consecuencia de las características sideróforas de su cadena lateral, que complementa a la habitual difusión pasiva a través de las porinas de la membrana externa del microorganismo de las cefalosporinas. En todo caso, su mecanismo de acción farmacológica es el propio de los fármacos de ese grupo –de todos los antibióticos betalactámicos–, o sea, la inhibición de la síntesis de la pared celular por unión a las proteínas de fijación de las penicilinas (PFP). En base a estas acciones, cefiderocol ha sido autorizado con indicación en el tratamiento por vía intravenosa y a nivel intrahospitalario de infecciones debidas a microorganismos gramnegativos aerobios en adultos con opciones terapéuticas limitadas, siguiendo siempre las guías oficiales relativas al uso correcto de antibacterianos.
Los datos clínicos que sustentaron su aprobación derivan de tres estudios pivotales, dos de fase 3 y otro de fase 2. En este último, el estudio APEKS-cUTI, la comparación de la eficacia del nuevo antibiótico frente a la asociación imipenem/cilastatina en pacientes con infecciones urinarias complicadas o pielonefritis no complicadas demostró su no inferioridad para la variable de curación en el día 7 de tratamiento, con una proporción superior de pacientes curados en el brazo de cefiderocol (73% vs. 55%). Sin embargo, la exclusión de pacientes con bacterias gramnegativas resistentes a carbapenemas impide realizar una comparación adecuada para establecer la utilidad de cefiderocol en infecciones causadas por este tipo de microorganismos.
En el caso del estudio de fase 3 CREDIBLE-CR, que analizó la eficacia de cefiderocol respecto a la mejor alternativa disponible, los resultados obtenidos en ambos brazos de tratamiento respecto a la proporción de pacientes con neumonía o sepsis que alcanzó la curación clínica en el día 7 tras el final del tratamiento fueron similares (aproximadamente 50% y 43%, respectivamente). En el caso de la erradicación microbiológica en pacientes con ITUc, también en el día 7 tras finalizar el tratamiento, la proporción fue superior en el brazo de cefiderocol (53% vs. 20%), pero el diseño descriptivo del estudio no permite concluir sobre la significación de estos resultados. Además, el número de pacientes finalmente incluido en cada subgrupo de tratamiento fue muy reducido, lo que limita aún más la posibilidad de extraer conclusiones respecto a la eficacia de cefiderocol. También resulta alarmante el dato de mortalidad, superior en el brazo de cefiderocol (34% vs. 18%), especialmente en los pacientes con infección por A. baumannii y que podría relacionarse con una baja actividad antibiótica de cefiderocol frente a este microorganismo o con fenómenos de heterorresistencia ocurridos durante el tratamiento. La mortalidad observada en infecciones causadas por otros patógenos fue similar a la de otros tratamientos ya disponibles (AEMPS, 2023).
Por último, el tercer ensayo pivotal, el estudio de fase 3 APEKS-NP, analizó como variable principal la eficacia de cefiderocol en comparación con meropenem en la reducción de la mortalidad por cualquier causa en el día 14 tras el inicio del tratamiento de neumonías bacterianas agudas. Los resultados fueron similares en ambos brazos de tratamiento, en torno al 12%, que indican la no inferioridad de cefiderocol. Estos resultados, semejantes en ambos brazos de tratamiento, podrían avalar el uso de cefiderocol cuando otras alternativas no estén disponibles o estén desaconsejadas.
Desde el punto de vista de la seguridad, el perfil toxicológico del nuevo fármaco parece aceptable, en la línea de lo que podría esperarse por lo conocido para otras cefalosporinas de última generación. Los eventos adversos observados en los ensayos clínicos fueron similares a los de los comparadores activos y las reacciones asociadas al tratamiento (diarrea, rash cutáneo, reacciones de hipersensibilidad, disminución del umbral convulsivo y hepatotoxicidad) tiene habitualmente intensidad leve o moderada, conllevando la discontinuación del tratamiento solo en muy raras ocasiones.
Las limitaciones en los datos disponibles sobre la eficacia clínica de cefiderocol han llevado a la EMA (2020) a considerar los datos de los estudios in vitro, del análisis de farmacocinética/farmacodinamia y las simulaciones de probabilidad de éxito decisivos a la hora de emitir una opinión positiva en la evaluación de este nuevo fármaco.
A pesar de los prometedores resultados de cefiderocol en los estudios preclínicos, los datos de eficacia obtenidos en ensayos clínicos ponen en entredicho su potencial utilidad. Cefiderocol plantea una novedosa forma de entrada a las bacterias gramnegativas que podría permitir solventar algunos de los mecanismos habituales de resistencia en estos microorganismos, pero no es descartable que en algunos casos se pueda producir resistencia de manera rápida, incluso intra-tratamiento. En base a los resultados obtenidos en los ensayos clínicos y preclínicos, la utilidad de cefiderocol podría ser superior en patógenos productores de metalo-betalactamasas, para los que las alternativas terapéuticas son muy limitadas.
Por tanto, aunque se requiere de resultados más exhaustivos procedentes de la práctica clínica habitual, no parece que cefiderocol vaya a constituir una alternativa disruptiva en infecciones por bacterias gramnegativas multirresistentes, más allá de los casos en que otros tratamientos para los que se disponga de mayor experiencia hayan fracasado (por resistencias) o estén desaconsejados (por intolerancia).
Valoración