Resumen
Como viene siendo habitual, inauguramos el primer número anual de Panorama Actual del Medicamento con un resumen de todos los medicamentos con nuevos principios activos comercializados por primera vez en España en el último año. Continuando la tendencia que marcó el 2021, y a pesar de la imperante pandemia por COVID-19, el recién terminado 2022 ha sido un año bastante prolífico en cuanto a la incorpora- ción al arsenal terapéutico de innovaciones farmacológicas, habiéndose comercializado un total de 32 nuevos principios activos, solo 3 menos que el año previo, pero 21 más si se compara con el difícil 2020. Más de la mitad de los nuevos fármacos se enmarcan los grupos terapéuticos ATC L (14, terapia antineoplásica y agentes inmunomoduladores) y J (5, antiinfecciosos para uso sistémico); en línea con lo acontecido en el último lustro, la mayor parte de los nuevos principios activos se dirigen al tratamiento de patologías on- cológicas o de naturaleza autoinmune, si bien desde 2021 la autorización de medicamentos frente a la COVID-19 también ha hecho crecer en abundancia el grupo de los antiinfeccio- sos. En el último año, además, se han incorporado un número no desdeñable (7) de principios activos en medicamentos designados como huérfanos.
A nivel de volumen de medicamentos, hay que destacar que por cuarto año consecutivo se reduce el número de presentaciones de medicamentos comercializadas, pues en 2022 se han comercializado 1.144 nuevas presentaciones –tanto de nuevos principios activos como de los ya existentes– frente a las 1.892 que se han dado de baja. A finales del año, el mercado de medicamentos en España contaba con aproximadamente 18.800 formatos o presentaciones comerciales de medicamentos. En la última década se han incorporado 9.557 presentaciones, lo que supone un 51% del total disponibles, y han desaparecido 11.243, con un balance negativo de 1.202 formatos solo en los últimos 3 años, lo cual refleja la tendencia a la renovación del mercado de medicamentos.
En cuanto al grado de innovación terapéutica, destaca sobremanera la comercialización y uso en España de la primera opción para el tratamiento por vía oral de la COVID-19 leve-moderada (Paxlovid®), que ofrece una ventana de oportunidad importante para prevenir casos graves, hospitalizaciones y muertes por la enfermedad. Adicionalmente, se ha comercializado lumasirán, un nuevo ARN pequeño de interferencia autorizado por vía subcutánea en pacientes con hiperoxaluria primaria de tipo 1 (HP-1) y que representa el primer tratamiento dirigido frente a la fisiopatología específica de la enfermedad. También merece una mención la comercialización de cabotegravir, el primer antirretroviral incluido en un medicamento inyectable, el cual presenta una posología muy ventajosa en comparación con otros tratamientos estándar y podría tener gran potencial en la profilaxis pre-exposición.
Las principales características fármaco-clínicas de todos los nuevos principios activos de 2022 se resumen en el presente artículo.
________________________________________________________________________
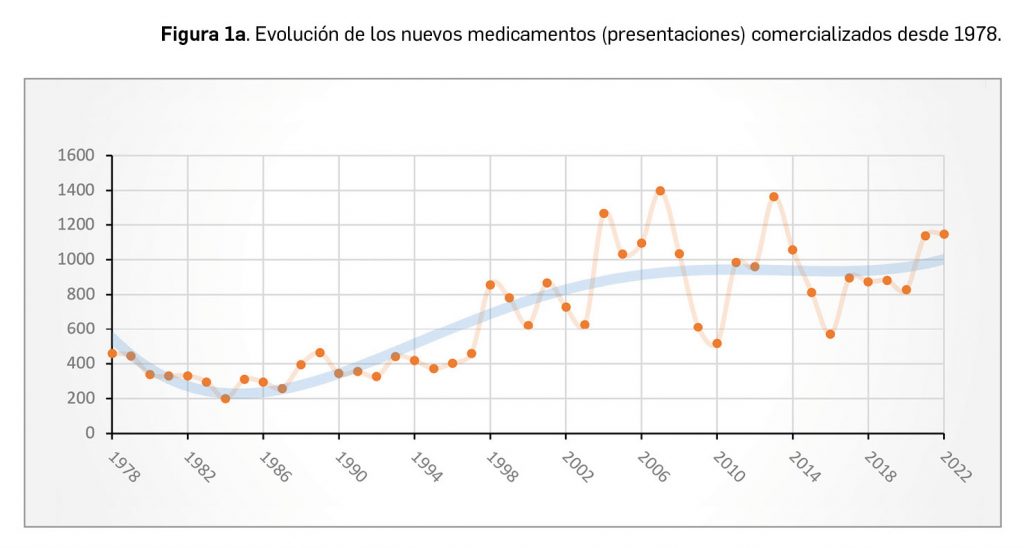
Durante el año 2022, se registraron en España 1.144 nuevas presentaciones comerciales o formatos de medicamentos, tal y como se recoge en la Tabla 1, incluyendo el 90% de ellas principios activos previamente comercializados. El dato se alinea con la tendencia mostrada en los periodos analizados, en los últimos 45 años (Figura 1a) y en los últimos 10 años (Figura 1b).
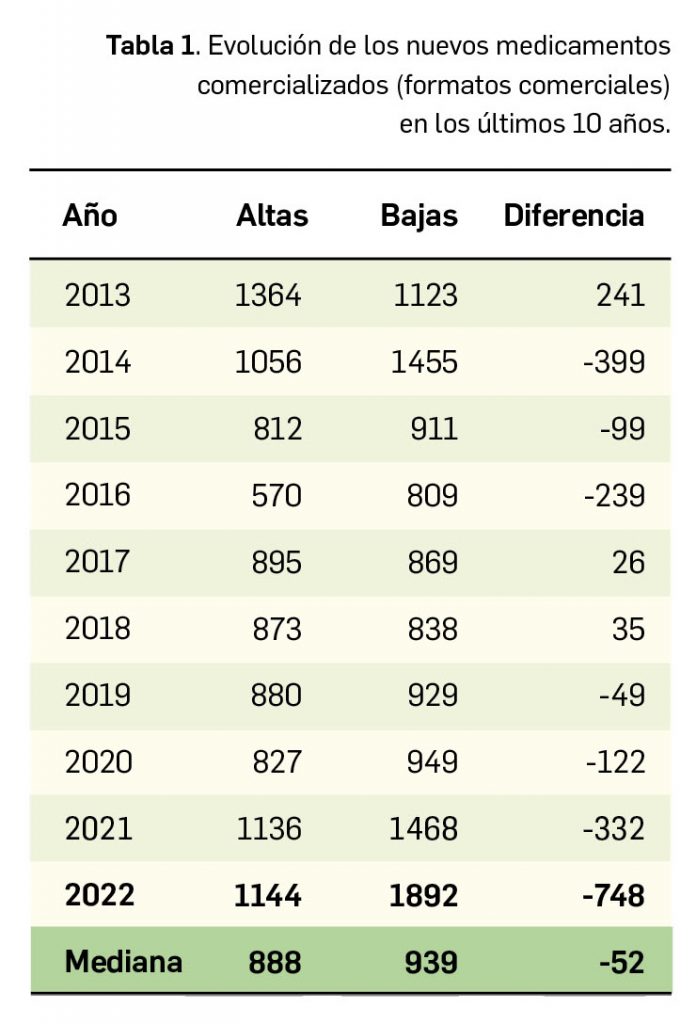
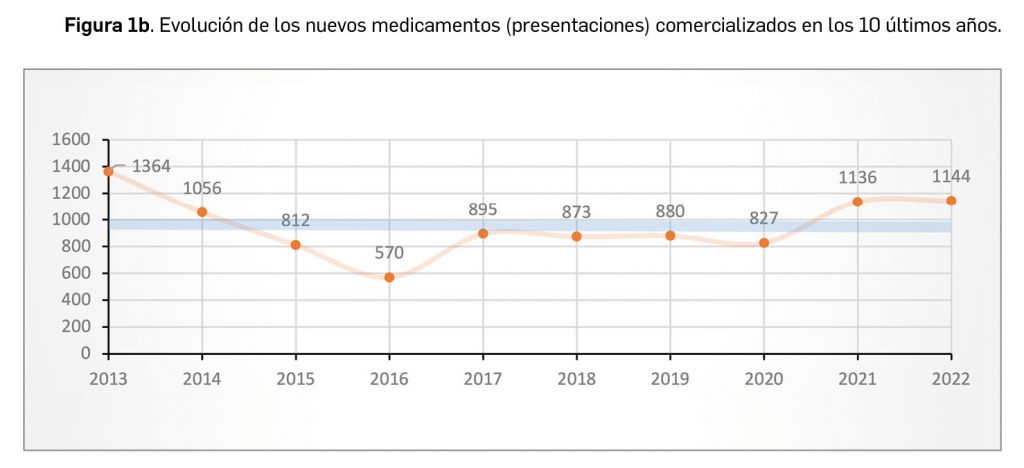
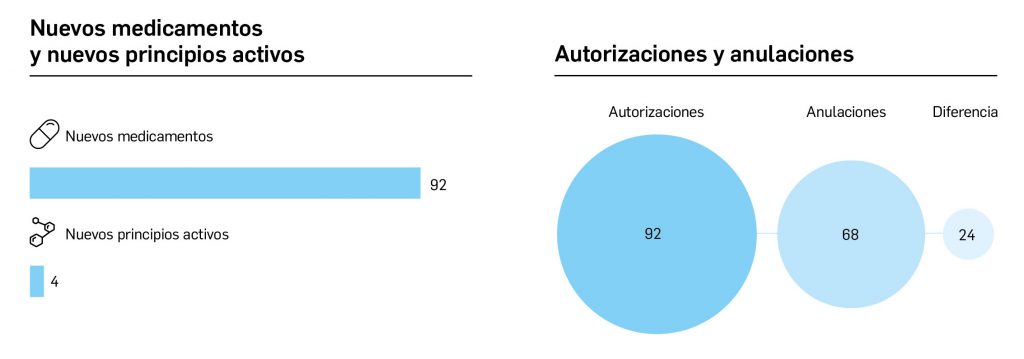
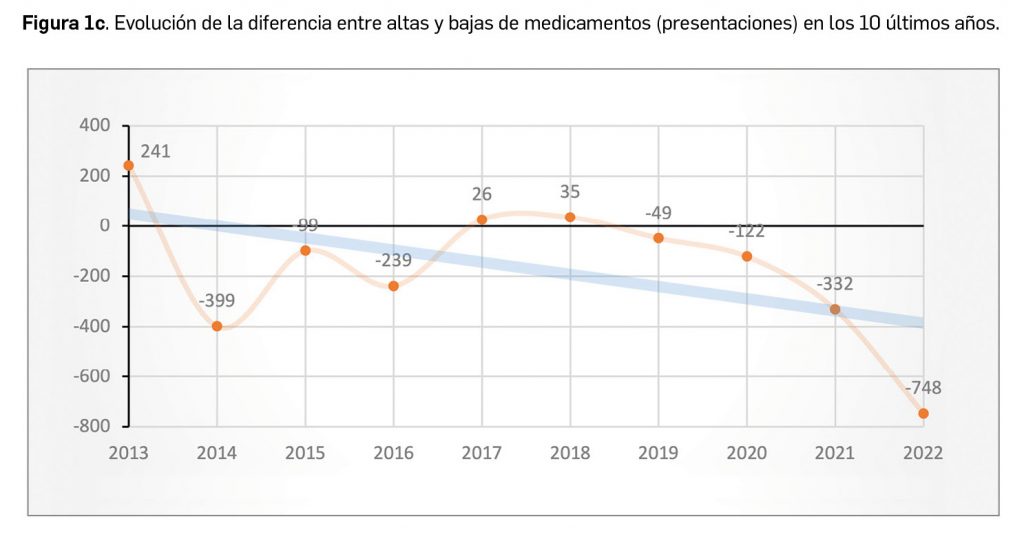
Cabe destacar, por ejemplo, que un total de 686 presentaciones corresponden a medicamentos genéricos, lo cual representa el 60% del total de nuevas presentaciones. Sin embargo, en el último año también se dieron de baja o anularon un total de 1.892 presentaciones, lo que arroja el mayor balance negativo hasta ahora registrado en el número de medicamentos autorizados. A fecha de 31 de diciembre de 2022, se encontraban en situación de comercialización un total de 18.779 presentaciones comerciales de medicamentos. Por otro lado, durante los últimos 10 años se han incorporado al mercado 9.557 presentaciones, lo que supone el 50,9% del total disponible actualmente; no obstante, también desaparecieron 11.243 presentaciones, lo cual se traduce en un descenso neto de 1.686 presentaciones en ese periodo. La tendencia a la renovación viene determinada por el incremento del número de bajas respecto a periodos anteriores, que supera con creces al número de altas en los últimos 3 años, en los que ha habido un total de 1.202 más bajas que altas (Figuras 1b y 1c).
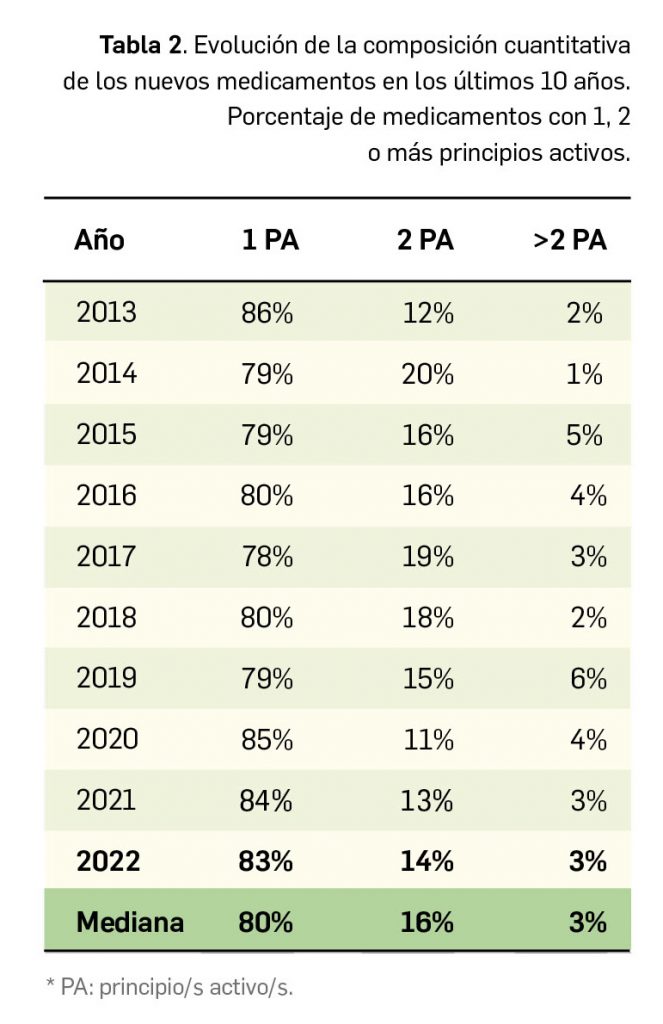
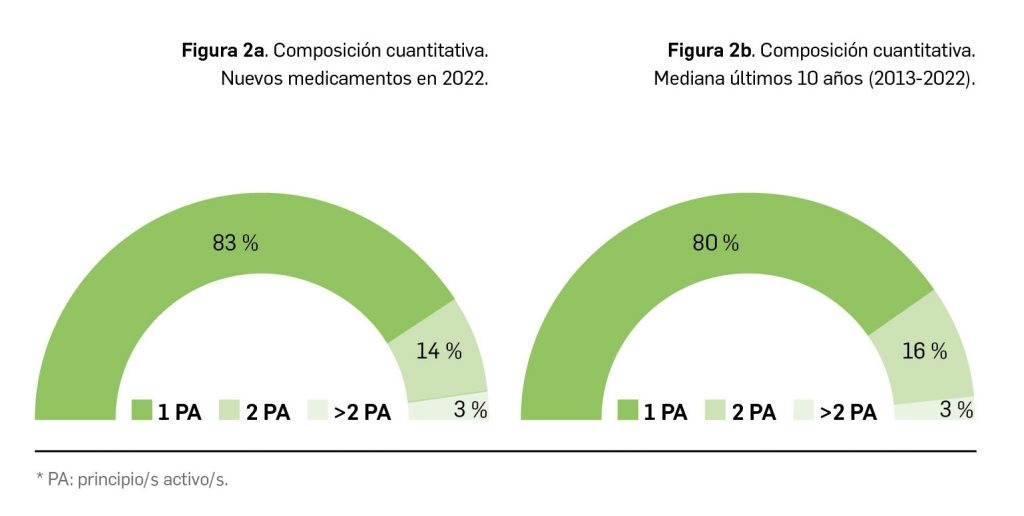
En lo relativo a la composición cuantitativa de los nuevos medicamentos comercializados en 2022, el 83% de estos fueron monocomponente, un 14% contenían dos principios activos y el restante 3% eran medicamentos multicomponente (Figura 2a). En este sentido, parece evidente que se mantiene la tendencia hacia los medicamentos monocomponente, que suponen el 80% de los nuevos medicamentos comercializados en los últimos 10 años (Tabla 2 y Figura 2b).
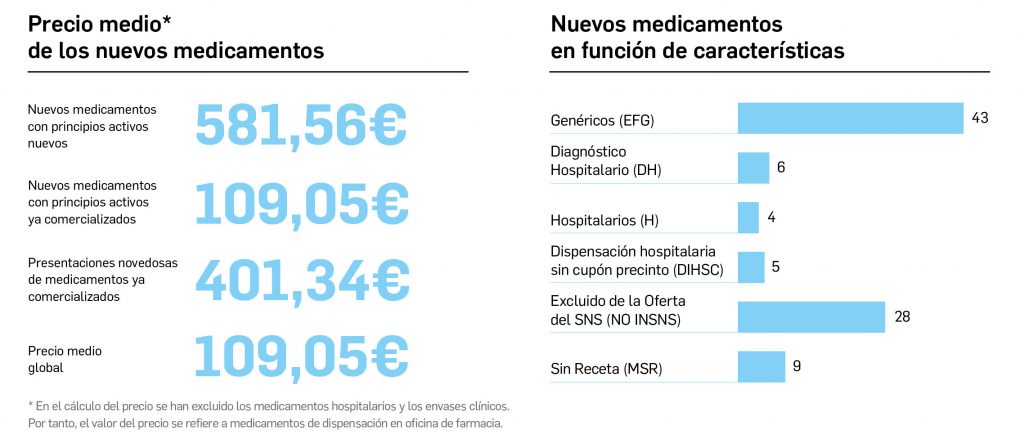
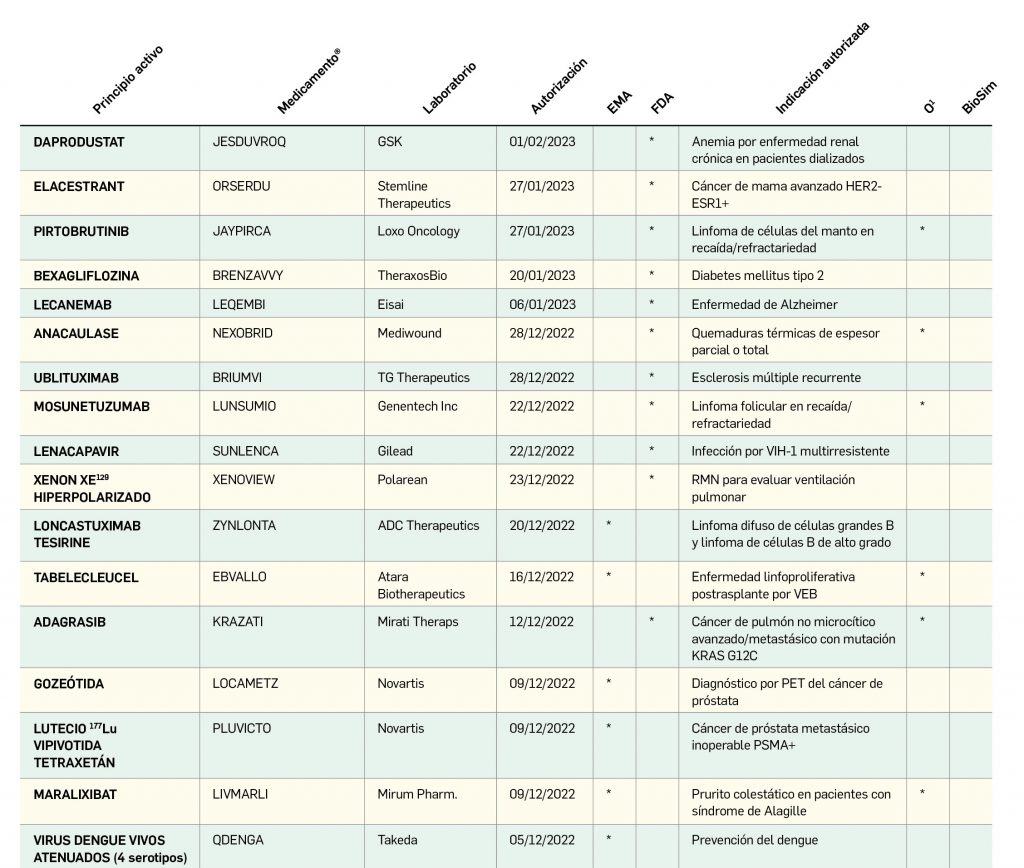
Por lo que se refiere a los nuevos principios activos comercializados en 2022 en nuestro país, han sido un total de 32 –de los cuales hasta 7 han sido incluidos en medicamentos designados como como huérfanos (un 22%)– (Tabla 3), una cifra muy similar a la constatada durante el prolífico 2021 y que se enmarca como normal en la tendencia de la última década (2013-2022), en que la mediana es de 30 nuevos principios activos/año, mientras que el promedio de nuevos principios activos/año se sitúa en 27,1. Teniendo en cuenta que se han comercializado un total de 1.144 presentaciones de medicamentos en 2022, se obtiene un ratio de 35,8 presentaciones nuevas de medicamentos por cada nuevo principio activo comercializado1, solo ligeramente superior a la mediana de la última década (29,6). En este sentido, cabe destacar que el 90% de los nuevos medicamentos comercializados durante 2022 incluyeron principios activos autorizados y comercializados en años previos; de ellos, un 3% son presentaciones novedosas de medicamentos ya comercializados previamente, entendiendo como tal aquellas que suponen una innovación en forma farmacéutica y/o vía de administración.
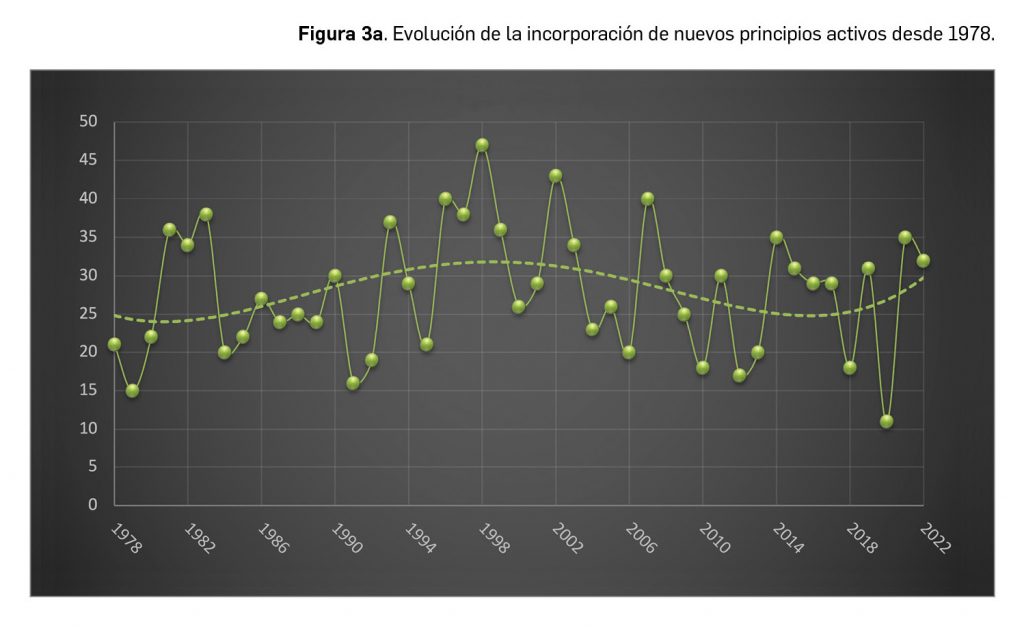
A grandes rasgos, las tendencias de la incorporación de nuevos principios activos en los últimos 45 años (1978-2022) (Figura 3a) y de la relación nuevas presentaciones/nuevos principios activos (Figura 3b) son moderadamente fluctuantes2. En los últimos 10 años se aprecia una ligera estabilización en la incorporación de nuevos principios activos en el mercado español (Figura 3c), que fue solo interrumpida en el año 2020 por la emergencia sanitaria mundial ocasionada por la COVID-19, cuando se produjo la menor comercialización de principios activos nuevos desde que se tienen registros en PAM, pero que se recuperó notablemente en 2021. El pasado año la relación global nuevas presentaciones/nuevos principios activos (35,8) se mantuvo nuevamente en valores muy cercanos a la mediana de la última década (29,6).

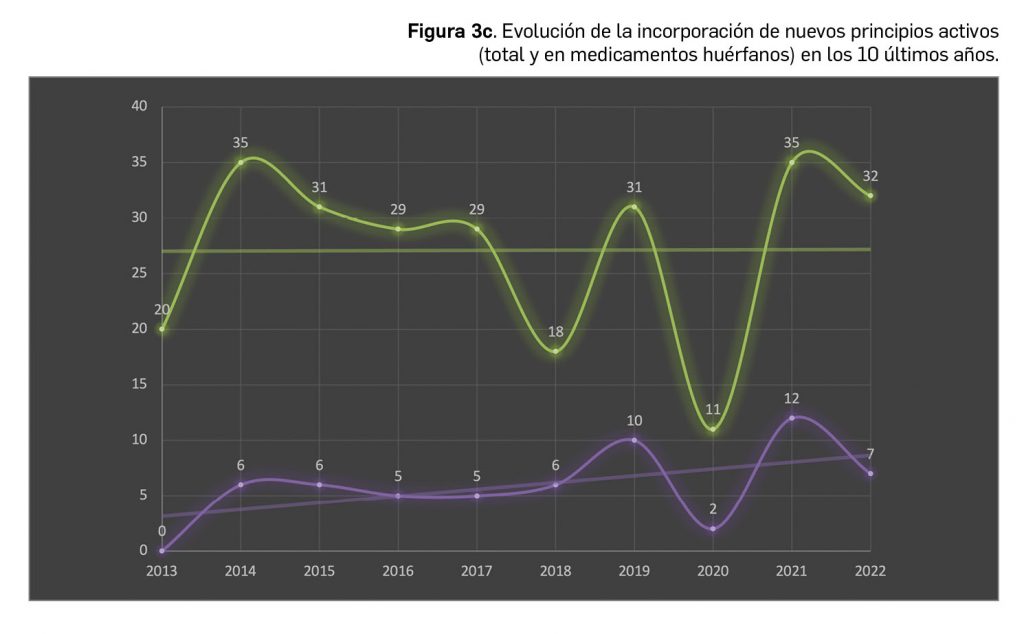
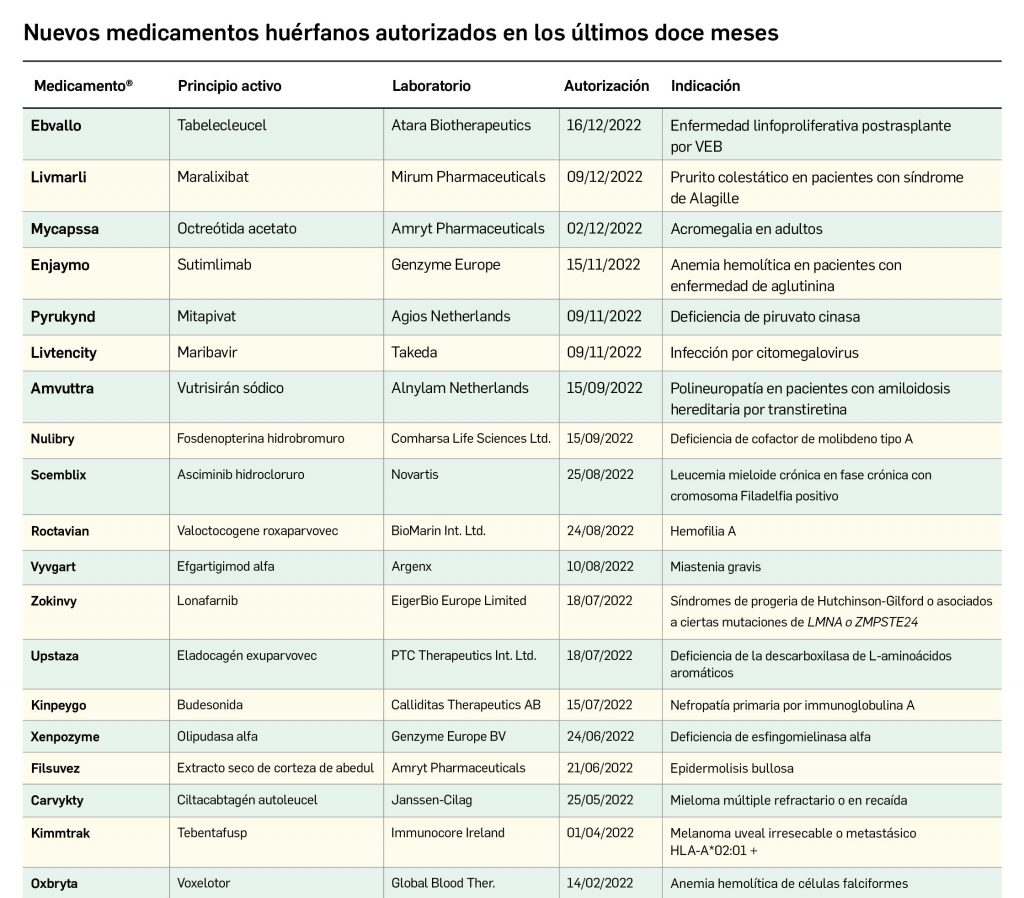
Desde el año 2002, se han comercializado en España 112 nuevos principios activos como medicamentos huérfanos3, lo que supone un 19,4% de los nuevos principios activos incorporados en ese periodo (un total de 577). La tendencia fue ligeramente ascendente hasta 2019 gracias a los 32 nuevos medicamentos huérfanos incorporados en los 5 años previos (2015-2019), pero también se vio interrumpida en 2020 por los solo 2 nuevos principios activos incluidos en medicamentos que habían sido designados como huérfanos; tras recuperarse igualmente con fuerza en 2021, la comercialización de este tipo de medicamentos mantiene un buen ritmo en este 2022, con 7 nuevos huérfanos. En cualquier caso, parece mantenerse cierta proporcionalidad entre el número total de nuevos principios activos y el de los incluidos específicamente en medicamentos huérfanos en cada año, lo que queda reflejado en el paralelismo entre ambas líneas de tendencia (Figura 3c).
Por otro lado, resulta reseñable que en el año 2022 solo se comercializó en España un nuevo medicamento biosimilar, concretamente de adalimumab (Yuflyma®). No obstante, se espera un crecimiento sustancial en la disponibilidad de biosimilares en un futuro cercano, ante la próxima expiración de las patentes de un número importante de medicamentos biológicos.
Por último, si se considera la clasificación terapéutica ATC de los nuevos principios activos comercializados en 2022, se han incorporado principios activos a 10 de los 14 grupos terapéuticos existentes. El grupo con mayor número de nuevos principios activos durante el año ha sido, como ya venía ocurriendo en los últimos años, el grupo L (terapia antineoplásica y agentes inmunomoduladores), con un total de 14. También cabe destacar, en un año en que la pandemia por COVID-19 se ha mantenido muy activa, la comercialización de 5 nuevos medicamentos enmarcados en el grupo J (antiinfecciosos para uso sistémico).
A modo de resumen, desde el año 1977, en que apareció por vez primera Panorama Actual del Medicamento, se han incorporado un total de 1.277 nuevos principios activos al mercado farmacéutico español, con independencia de su clasificación terapéutica.
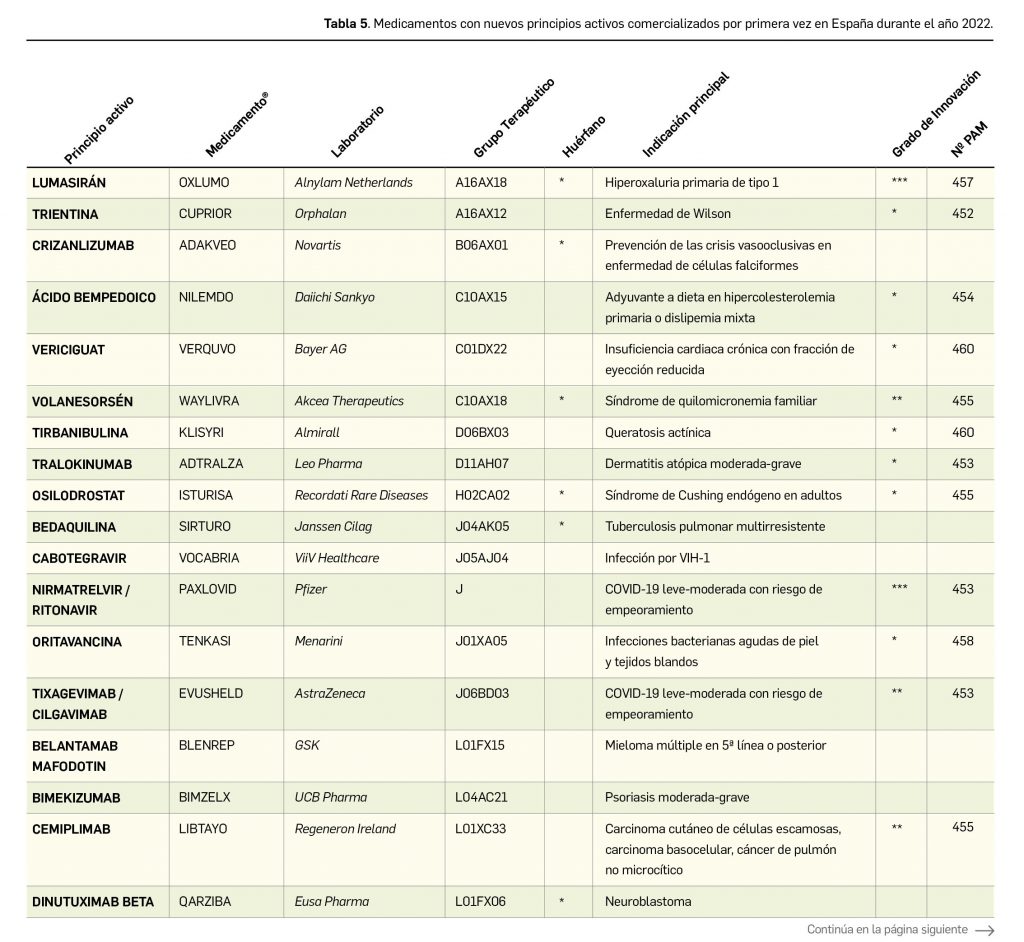
En la siguiente tabla (Tabla 5) se muestran los nombres de los nuevos principios activos comercializados por primera vez en España durante el año 2022, junto a los nombres de los medicamentos en que se incluyen, su grupo terapéutico ATC e indicación principal. Seguidamente, se resumen las principales características fármaco-clínicas de cada uno de ellos, clasificados por grupos ATC, en base a la información disponible en el momento de su primera comercialización. Para algunos de los medicamentos consignados en la tabla no se incluyen todavía sus resúmenes, los cuales aparecerán en los próximos números de Panorama Actual del Medicamento.
Tracto alimentario y metabolismo
HIPEROXALURIA PRIMARIA DE TIPO 1
LUMASIRÁN
▼OXLUMO® (Alnylam Netherlands) PAM 457
Lumasirán es un nuevo ARN pequeño de interferencia bicatenario que se dirige específicamente a los hepatocitos, donde se une al ARNm del gen HAO1 codificante para la enzima hidroxiácido oxidasa 1 o glicolato oxidasa y promueve su degradación mediante el proceso de interferencia del ARN. Por tanto, reduce los niveles de síntesis de esa proteína y provoca una disminución de la cantidad de glioxilato disponible: este es un sustrato de la producción de oxalato, de modo que también reduce los niveles de oxalato en orina y en plasma. Designado como huérfano, el medicamento ha sido autorizado para el tratamiento por vía subcutánea de la hiperoxaluria primaria de tipo 1 (HP-1) en todos los grupos de edad.
En un estudio pivotal (N= 39) con pacientes de > 6 años y función renal relativamente conservada, el tratamiento con lumasirán durante 6 meses fue superior a placebo en la tasa de reducción significativa de los niveles urinarios promedio de oxalato (65% vs. 12%). Así, el 52% y 84% de los pacientes tratados con lumasirán consiguen niveles de oxalato en orina normales o próximos a la normalidad, respectivamente. El tratamiento se relacionó, además, con una reducción del 40% en los niveles de oxalato en plasma, que apenas cambiaron con placebo. Los resultados de un segundo estudio de fase 3 con pacientes pediátricos de < 6 años (N= 18) respaldan esos hallazgos, pues el tratamiento abierto durante 6 meses con lumasirán redujo el cociente oxalato:creatinina en orina un promedio del 72% respecto al nivel basal, y la mitad de los pacientes alcanzó un valor casi normal; además, también se vio un descenso del 32% en el nivel plasmático de oxalato, con estabilidad en la funcionalidad renal durante ese periodo. En términos de seguridad, se trata de un fármaco bien tolerado cuyo perfil toxicológico, similar en adultos y en niños, se caracteriza por la incidencia de eventos adversos (42% vs. 8% con placebo) leves-moderados que no conllevan una tasa reseñable de interrupciones: las reacciones adversas más frecuentes son las del sitio de inyección (25%, sobre todo eritema, dolor y prurito transitorios) y el dolor abdominal (15%).
Bien es cierto que la eficacia de lumasirán, que es rápida y parece mantenerse en tratamientos de al menos 1 año, se ha evaluado a través de variables bioquímicas intermedias; no se tienen datos de variables de mayor relevancia clínica –calidad de vida o mortalidad, por ejemplo– o en pacientes con enfermedad renal grave, pero es bastante plausible que tales resultados se correlacionen con una reducción de los depósitos extrarrenales de oxalato y menor incidencia de comorbilidades asociadas a HP-1, e incluso que se traduzcan en una reducción de la progresión de la patología. Pero por ahora no se ha observado un beneficio a nivel de eventos renales ni de la funcionalidad renal, probablemente debido al pequeño tamaño muestral y los cortos periodos de seguimiento.
En ausencia de datos a largo plazo y de comparaciones directas o indirectas entre lumasirán y las otras opciones usadas en práctica clínica (hiperhidratación, inhibidores de la cristalización o piridoxina), no se puede concluir sobre su superioridad, pero es evidente que el nuevo fármaco inaugura una vía terapéutica en una indicación para la que no se disponía de tratamientos farmacológicos aprobados en la UE y puede cambiar en un futuro la terapéutica estándar de forma notable. No representa una cura, pero permite combatir las manifestaciones clínicas de la HP-1 debidas a la acumulación de oxalato, posicionándose como una alternativa terapéutica en aquellos pacientes sin antecedentes de trasplante y con enfermedad renal leve o moderada que no responden a las opciones de tratamiento estándar. La evidencia es aún escasa, y habrá que esperar a los procedentes de los ensayos en marcha para definir mejor su balance beneficio-riesgo a largo plazo.
ENFERMEDAD DE WILSON
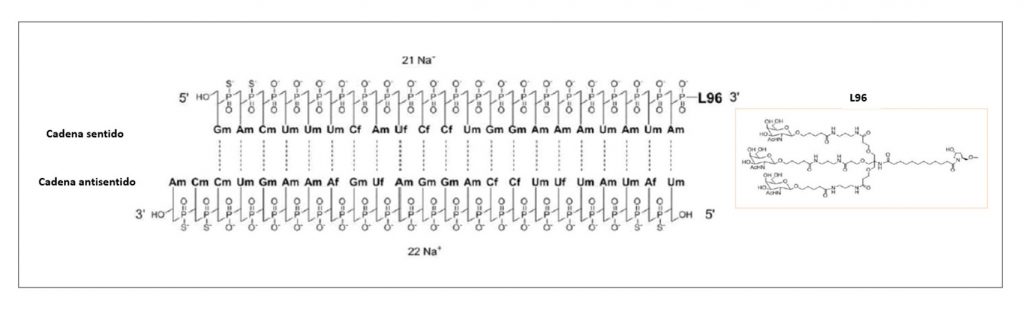
TRIENTINA
▼CUPRIOR® (Orphalan) PAM 452
La trientina es un quelante selectivo del cobre que potencia la eliminación sistémica del cobre divalente libre al formar con él un complejo estable fácilmente excretado por orina; además, puede quelar el cobre en el intestino e inhibir su absorción. Esa acción dual resultará en niveles sistémicos estables o reducidos de cobre libre y permitirá controlar las complicaciones multiorgánicas debidas al exceso patológico de este metal. El nuevo medicamento ha sido autorizado para el tratamiento por vía oral de la enfermedad de Wilson (EW) en adultos, adolescentes y niños de ≥ 5 años de edad que no toleran el tratamiento con D-penicilamina.
El fármaco se ha usado, en distintas formas, durante > 30 años para tratar la EW, por lo que a pesar de que es nuevo principio activo en España no dispone de IPT. La autorización del medicamento con la sal tetrahidrocloruro se ha sustentado en un dossier basado en una revisión bibliográfica de las propiedades fármaco-toxicológicas de la trientina. Así, la extrapolación de los datos farmacocinéticos respecto a trientina dihidrocloruro (disponible en países como Reino Unido) se basó en un ensayo comparativo que permitió concluir que la principal diferencia es que la sal tetrahidrocloruro “libera” más cantidad de fármaco activo, lo cual puede contrarrestarse con un simple ajuste de dosis y la medición de los niveles orgánicos de cobre. La eficacia y seguridad clínicas se extrapolaron a partir de los resultados de un estudio de cohortes retrospectivo (estudio Lariboisière), que comparó el uso de ambas sales en 43 pacientes con EW en programas de uso compasivo de un solo hospital. Los síntomas hepáticos mejoraron en el 23% de los tratamientos con trientina tetrahidrocloruro y en el 30% de aquellos con la sal dihidrocloruro, y los síntomas neurológicos, en el 31% y el 27%, respectivamente. En general, ambos tratamientos indujeron desde el inicio una mejora en los niveles séricos de transaminasas hepáticas y una estabilización o reducción de los de cobre libre en sangre y orina, sin diferencias significativas entre las dos sales de trientina.
Ninguno de los pacientes que recibió el fármaco presentó eventos adversos relacionados con el tratamiento, ni hubo muertes asociadas. Se acepta que el fármaco tiene un perfil toxicológico benigno, en el que la reacción adversa más frecuente será la aparición de náuseas al principio del tratamiento; también pueden surgir algunas reacciones adversas cutáneas (erupción, prurito) o casos de deterioro neurológico inicial. Cualitativamente, sobresale el riesgo de posibles casos graves de anemia ferropénica y de colitis. De igual modo, habrá que valorar cuidadosamente el beneficio-riesgo en embarazadas y lactantes, y tener presente su potencial de interaccionar con alimentos y otros medicamentos en intestino.
En definitiva, los datos disponibles sugieren que el tratamiento de la EW con trientina tetrahidrocloruro puede ser mejor tolerado, pero menos eficaz que con penicilamina. Sin novedad relevante en el plano mecanístico respecto a esta, el “nuevo” fármaco ha sido además aprobado en una 2ª línea de tratamiento posterior al uso de penicilamina. Es decir, será relevante para los pacientes que no responden a la 1ª línea, pero se trata de un tratamiento sintomático, no curativo, que se establecerá por periodos prolongados y no parece aportar ningún elemento sustancial de innovación terapéutica en la enfermedad de Wilson.
Sistema cardiovascular
HIPERCOLESTEROLEMIA PRIMARIA O DISLIPEMIA MIXTA
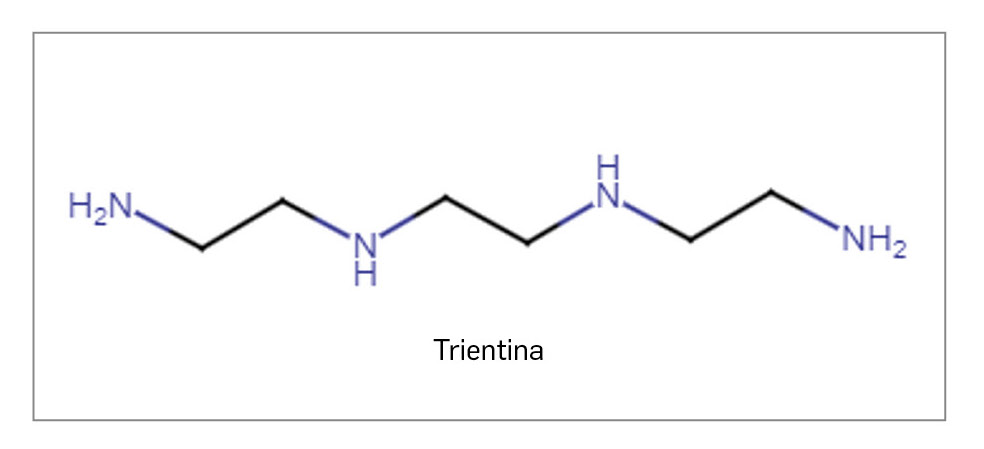
ÁCIDO BEMPEDOICO
▼NILEMDO® (Daiichi Sankyo) PAM 454
El ácido bempedoico es un nuevo profármaco que, al activarse por unión a la coenzima A, actúa como inhibidor competitivo de la enzima adenosina trifosfato-citrato liasa, de modo que inhibe la síntesis de colesterol en el hígado y reduce sus niveles intracelulares, así como los de colesterol-LDL en plasma. El medicamento ha sido autorizado para el tratamiento diario por vía oral de pacientes adultos con hipercolesterolemia primaria (familiar heterocigótica y no familiar) o dislipidemia mixta, como adyuvante a la dieta, bien en combinación con una estatina sola o con una estatina y otros tratamientos hipolipemiantes en pacientes que no puedan alcanzar sus objetivos de c-LDL con la dosis máxima tolerada de una estatina; también se ha aprobado en monoterapia o en combinación con otros tratamientos hipolipemiantes en pacientes intolerantes o con contraindicación al uso de estatinas.
En un total de 4 ensayos pivotales de adecuado diseño (N> 3.600) ha demostrado un efecto hipolipemiante moderado. En pacientes con alto riesgo cardiovascular, su asociación a dosis máximas de estatinas redujo significativamente los niveles de c-LDL a las 12 semanas de tratamiento, en mayor medida que placebo (diferencia de -18%)2, y permitió a una mayor proporción de pacientes llegar al objetivo de < 70 mg/dl (29% vs. 8%); también probó su eficacia en este grupo de pacientes en combinación con dosis fijas con ezetimiba (reducción de c-LDL en -38% de diferencia con placebo). En pacientes intolerantes a las estatinas, la administración de ácido bempedoico indujo un descenso sustancial en los niveles medios de c-LDL a los 3 meses, con un -25% de diferencia respecto a placebo. Además, redujo globalmente los niveles de colesterol no HDL, de apolipoproteína B y de colesterol total, con una eficacia consistente en todos los subgrupos de pacientes, que se mostró máxima tras 1 mes y se mantuvo en niveles similares durante periodos de al menos 1 año. En términos de seguridad, se trata de un fármaco con un perfil toxicológico relativamente benigno y manejable clínicamente. La incidencia de reacciones adversas es baja y similar al uso de placebo, siendo en su mayoría leves-moderadas, por lo que la tasa de interrupción del tratamiento es también baja. Sobresalen por su frecuencia (< 5%) las siguientes reacciones adversas: hiperuricemia, mialgias, espasmo muscular, diarrea y cefalea. Se ha descrito una mayor incidencia de alteraciones hepáticas y renales, pero no parecen tener una gran relevancia clínica.
No se ha confirmado aún la eficacia del fármaco sobre la incidencia de eventos cardiovasculares y la morbi-mortalidad asociada. Ese objetivo clínico sí ha sido probado, en cambio, con el uso de estatinas, ezetimiba y los inhibidores de PCSK9 (evolocumab y arilocumab); las comparaciones indirectas con estos últimos apuntan a una inferioridad del ácido bempedoico. Tampoco se conoce su balance beneficio-riesgo en tratamientos de más de 1 año, por lo que habrá que esperar a los resultados de los estudios ahora en marcha para sacar conclusiones sólidas. En definitiva, pese a que incorpora un novedoso mecanismo de acción y es bien tolerado, el beneficio clínico esperable con el nuevo fármaco es modesto. Su adición a la terapia intensiva con estatinas y ezetimiba permitiría rescatar a un 15-20% de pacientes de alto riesgo adicionales, si los niveles de c-LDL están < 20% por encima del nivel objetivo; y en pacientes intolerantes a estatinas, su uso junto a ezetimiba podría favorecer una reducción de c-LDL un 20-30% adicional (insuficiente en la mayoría de pacientes de alto riesgo). El ácido bempedoico se posicionará, pues, como una alternativa terapéutica en tercera línea de tratamiento, sobre todo en pacientes no candidatos al uso de inhibidores de PCSK9. No parece incorporar cambios sustanciales en la terapéutica estándar de la hipercolesterolemia.
INSUFICIENCIA CARDIACA
VERICIGUAT
▼VERQUVO® (Bayer) PAM 460
Se recomienda consultar el artículo monográfico sobre vericiguat que se publica en este mismo número.
SÍNDROME DE QUILOMICRONEMIA FAMILIAR
VOLANESORSÉN
▼WAYLIVRA® (Akcea Therapeutics) PAM 455
Volanesorsén es un nuevo oligonucleótido antisentido diseñado específicamente para unirse al ARNm e inhibir la síntesis de la apolipoproteína C-III (apoC-III), de modo que favorece el aclaramiento hepático de los quilomicrones y otras lipoproteínas ricas en triglicéridos. Designado como huérfano, el medicamento ha recibido la autorización condicional como complemento a la dieta en pacientes adultos con síndrome de quilomicronemia familiar (SQF) confirmado genéticamente y con riesgo alto de pancreatitis, en quienes la respuesta a la dieta y al tratamiento de reducción de triglicéridos no ha sido suficiente. Esa combinación era, precisamente, la única terapia hasta ahora viable en el tratamiento a largo plazo del SQF, con eficacia limitada.
En un estudio pivotal de fase 3, un tratamiento semanal indujo a los 3 meses una reducción significativa de -77% en los niveles de triglicéridos respecto al estado basal (vs. +18% con placebo; diferencia de -94%). La práctica totalidad de pacientes responden al tratamiento y una gran parte alcanza el objetivo de trigliceridemia de < 750 mg/dl (77% vs. 10% con placebo). Con un inicio rápido, el efecto reductor de triglicéridos del fármaco se atenúa con el tiempo de tratamiento (diferencia de -53% a los 6 meses y -40% a los 12 meses), lo cual puede deberse al menos en parte a la mayor tasa de interrupciones del tratamiento por motivos de seguridad. La superior eficacia del fármaco se mostró consistente con independencia del uso de hipolipemiantes y fue respaldada por los datos del estudio abierto de extensión, también en tratamientos de más de 1 año. Por otra parte, en su perfil toxicológico –manejable clínicamente– sobresale el riesgo de trombocitopenia (> 30%), que, aunque suele ser leve-moderada y tratable y no se asocia con hemorragias graves, determina a menudo la interrupción del fármaco. Junto a la alta incidencia de reacciones locales en el punto de inyección (> 80%), otros efectos adversos frecuentes durante el tratamiento son: cefalea, fatiga, dolor abdominal, diarrea o náuseas. Aún persisten incertidumbres sobre la relevancia de su inmunogenicidad y la seguridad –y eficacia– en mayores de 65 años.
En resumen, volanesorsén inaugura una ruta farmacológica en su indicación, siendo el primer fármaco específicamente autorizado frente al síndrome de quilomicronemia familiar; continúa la idea terapéutica de los nucleótidos antisentido iniciada en 2018 por nusinersén (usado en atrofia muscular espinal), aunque tiene mejor perfil de seguridad de lo que sería esperable en este tipo de terapias génicas (no provoca toxicidad hepática o renal sustancial). No obstante, las limitaciones de la evidencia disponible (relativas al uso de un variable subrogada o a la representatividad de la población de participantes) y las incertidumbres sobre su beneficio-riesgo a largo plazo (el efecto decae con el tiempo y hay una alta tasa de interrupciones), su utilidad real en práctica clínica en el uso crónico que requeriría el SQF puede verse seriamente comprometida, a falta de disponer de datos adicionales de efectividad y tolerabilidad.
Dermatológicos
QUERATOSIS ACTÍNICA
TIRBANIBULINA
▼KLISYRI® (Almirall) PAM 460
Se recomienda consultar el artículo monográfico sobre tirbanibulina que se publica en este mismo número.
PSORIASIS EN PLACAS
TRALOKINUMAB
▼ADTRALZA (Leo Pharma) PAM 453
Tralokinumab es un anticuerpo monoclonal humano que se une específicamente a un epítopo de la IL-13 que se superpone con su sitio de unión a los receptores α de IL-13 (IL-13Rα), y previene la unión de esta citocina tanto a los receptores IL-13Rα1/IL-4Rα como a los IL-13Rα2. Así, neutraliza la actividad biológica de la IL-13, que es, junto a la IL-4, una de las principales citocinas implicadas en la patogenia de la inflamación tipo 2: entre otras acciones, favorece el cambio de isotipo de las células B para la producción de IgE, el reclutamiento de células efectoras proinflamatorias o la alteración de la barrera epidérmica. Dado que la inhibición de su vía de señalización con tralokinumab tiene importantes efectos antiinflamatorios, el medicamento ha sido aprobado para el tratamiento por vía subcutánea de la dermatitis atópica (DA) de moderada a grave en adultos que son candidatos a tratamiento sistémico.
En una pauta quincenal, el nuevo fármaco ha demostrado una eficacia significativamente superior a placebo tanto en monoterapia como asociado a corticoides tópicos en tres estudios pivotales de adecuado diseño que han aleatorizado a casi 2.000 pacientes. Su uso en monoterapia durante 4 meses se asoció a tasas de aclaramiento total o casi total de la piel del doble de las obtenidas con placebo (respuesta según escala IGA 0/1 de 16-22% vs. 7-11%, y según EASI-75 de 25-33% vs. 11-13%); ese beneficio se corroboró con mantenimientos de hasta 1 año de duración. Combinado con corticoides tópicos la eficacia de tralokinumab a los 4 meses es incluso mayor: respuesta IGA 0/1 del 39% (vs. 26% con placebo) y de EASI-75 del 56% (vs. 36%), permitiendo reducir a la mitad el uso de la corticoterapia corticoides. Los resultados de las variables secundarias, como la intensidad del prurito o la calidad de vida asociada a DA, respaldan la eficacia significativa del nuevo fármaco, consistente en todos los subgrupos de pacientes (incluso en los no respondedores tras una fase inicial) y apreciable desde las semanas 1 o 2 de tratamiento. Los datos sugieren que la pauta mensual puede ser probablemente menos eficaz que el tratamiento con tralokinumab cada 2 semanas (régimen preferente).
A grandes rasgos, el fármaco es bien tolerado a medio plazo, con un perfil toxicológico definido y manejable. La incidencia de eventos adversos relacionados con el fármaco (14%) es similar a placebo, y la mayoría de estos son autolimitados y leves-moderados, lo cual determina una baja tasa de abandonos del tratamiento por motivos de seguridad (en torno al 2%). En la línea de otras proteínas terapéuticas frente a patologías con componente inflamatorio, con el uso de tralokinumab destacan por su frecuencia reacciones adversas como infecciones del tracto respiratorio superior (23%; mayoritariamente víricas, como resfriado común), reacciones en el punto de inyección (7%; como dolor y enrojecimiento) o conjuntivitis. El riesgo de reacciones alérgicas e inmunogenicidad parece bajo y sin relevancia clínica.
No parece que el nuevo fármaco incorpore un grado reseñable de novedad mecanística respecto al ya comercializado dupilumab (monoclonal dirigido a la subunidad α del receptor de IL-4), el cual inhibe tanto las vías de la inflamación tipo 1 (mediadas por la formación del dímero IL-4Rα/γc) como las de tipo 2 (mediadas por el dímero IL-4Rα/IL-13Rα). No se dispone de comparaciones directas entre ambos, ni tampoco entre tralokinumab y otros fármacos dirigidos de uso sistémico, como baricitinib o upadacitinib, pero las comparaciones indirectas sugerirían que tralokinumab no es superior a los anteriores. A falta de conocer las consideraciones del IPT, todo apunta a que el nuevo fármaco, también en investigación en asma, se posicionará como una alternativa a dupilumab en el tratamiento sistémico biológico de novo tras fracaso del tratamiento tópico en casos moderados-graves de DA, de especial interés en pacientes con respuesta inadecuada o intolerancia a ciclosporina. Dupilumab ya ha demostrado su efectividad en vida real y será probablemente la primera opción en ese contexto terapéutico hasta que se disponga de datos comparativos. Además, se requieren estudios a largo plazo que esclarezcan el beneficio-riesgo de tralokinumab con el uso prolongado que previsiblemente requerirá una enfermedad de curso crónico-recurrente como la DA.
Preparados hormonales sitémicos
SÍNDROME DE CUSHING ENDÓGENO
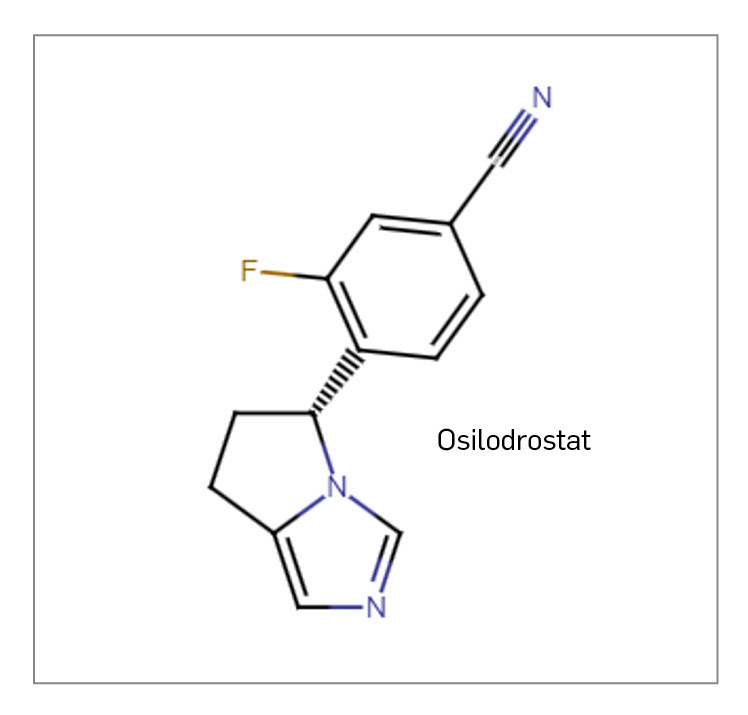
OSILODROSTAT
▼ISTURISA® (Recordati Rare Diseases) PAM 455
Osilodrostat es un nuevo inhibidor potente y selectivo de la 11β-hidroxilasa (CYP11B1), enzima responsable de la etapa final de la biosíntesis de cortisol en la glándula suprarrenal, de modo que reduce los niveles de cortisol sanguíneos; además, inhibe la acción de la enzima 18-hidroxilasa (CYP11B2), reduciendo la síntesis –y los niveles– de aldosterona. El medicamento, designado como huérfano, ha sido autorizado para el tratamiento por vía oral (dos veces al día) del síndrome de Cushing endógeno (SCE) en adultos.
Su autorización se ha sustentado en los datos clínicos de un ensayo pivotal de fase 3 (N= 137), en el que, a la semana 34, al final de la fase de retirada aleatorizada, la administración de osilodrostat aportó una respuesta completa –niveles de cortisol libre urinario en el rango de la normalidad– en el 86% de los pacientes; esta proporción casi triplica a la de pacientes con respuesta en el brazo con placebo (29%), lo que supone una probabilidad 14 veces mayor de alcanzar la respuesta (OR= 13,7; p< 0,001). El tiempo medio hasta la respuesta con el nuevo fármaco es de 41 días, manteniéndose la eficacia al menos hasta la semana 48 de tratamiento, cuando son respondedores dos tercios de los pacientes. También hubo una mejora de los signos y síntomas cardiovasculares, metabólicos y físicos relacionados con el síndrome de Cushing, así como en las escalas de calidad de vida. Por otra parte, el perfil toxicológico del fármaco, aunque clínicamente manejable con ajustes posológicos y en línea con lo descrito para otros fármacos aprobados en su indicación, es importante: todos los pacientes sufren al menos un evento adverso y los graves se consideran relacionados con el fármaco en el 30-40% de los pacientes; la tasa de discontinuación del tratamiento es del 10%. Los eventos adversos más frecuentes son los trastornos del tracto gastrointestinal (67-68%, sobre todo náuseas y vómitos), las infecciones (67-74%) y otras reacciones adversas inespecíficas (65-79%, incluidas fatiga, edema y cefalea). Más de la mitad de los pacientes notifican casos de hipocortisolismo (52%, mayoritariamente insuficiencia suprarrenal), quizás con mayor riesgo que otros anticorticosteroides, por lo que requerirá de controles periódicos. No obstante, se entiende que el perfil de eventos adversos se deberá principalmente a la propia enfermedad de base.
En ausencia de datos a largo plazo y de comparaciones directas con otras opciones aprobadas en el SCE (ketoconazol y metirapona), a pesar de que las comparaciones indirectas apuntan a una superioridad en eficacia, el nuevo fármaco se posiciona por ahora como como una opción más en la farmacoterapia del SCE, pero como alternativa tras el fracaso o contraindicación a ketoconazol o metirapona, balanceando los posibles eventos adversos según la situación del paciente. Dado que no incorpora una novedad reseñable en el plano mecanístico y que el tratamiento del síndrome de Cushing no incluye habitualmente la farmacoterapia en las primeras líneas (que serían la cirugía y la radioterapia), se comprende que osilodrostat no va a modificar sustancialmente la terapéutica estándar.
Antiinfecciosos de uso sistémico
INFECCIÓN POR SARS-CoV-2: COVID-19
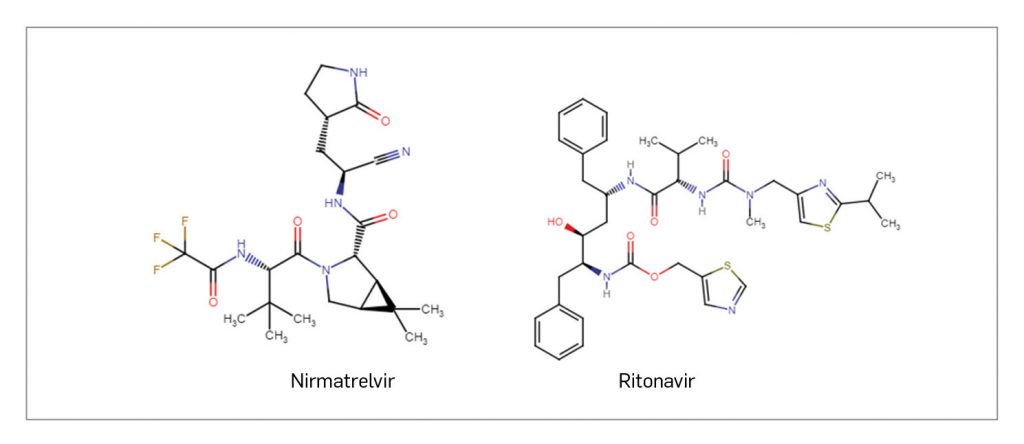
NIRMATRELVIR / RITONAVIR
▼PAXLOVID® (Pfizer) PAM 453
TIXAGEVIMAB / CILGAVIMAB
▼EVUSHELD® (AstraZeneca) PAM 453
Paxlovid® es una combinación del nuevo antiviral nirmatrelvir y del antirretroviral ya conocido ritonavir. El primero actúa como inhibidor peptidomimético potente y selectivo de la proteasa principal del SARS-CoV-2, bloqueando la capacidad del virus de procesar los precursores poliproteicos e impidiendo su replicación; parece que retiene actividad frente a las variantes virales más expandidas. Ritonavir, por su parte, actúa como potenciador farmacocinético: inhibe el metabolismo de nirmatrelvir por las isoenzimas hepáticas CYP3A4 y aumenta sus concentraciones plasmáticas. El medicamento ha recibido la aprobación condicional para el tratamiento de la COVID-19 por vía oral en adultos que no requieren aporte de oxígeno suplementario y que tienen un riesgo alto de progresar a enfermedad grave.
Se ha contrastado adecuadamente en un ensayo pivotal de fase 2/3 (N= 2.246) que el tratamiento, administrado en los primeros 5 días de manifestaciones en pacientes adultos con COVID-19 leve-moderada no vacunados ni convalecientes de la infección y con algún factor de riesgo de empeoramiento (edad avanzada, inmunodepresión o comorbilidades), reduce el riesgo de hospitalización por COVID-19 o muerte por cualquier causa en un 87% respecto a placebo. Con una eficacia consistente en todos los subgrupos de pacientes, y mayor en pacientes seronegativos, presenta un perfil toxicológico benigno (similar a placebo), en que la mayoría de los eventos adversos son leves-moderados; sobresalen por su frecuencia los trastornos del gusto o la diarrea. Se trata, pues, del primer antiviral de uso por vía oral frente a la COVID-19, que incorpora, además, una nueva diana terapéutica en la lucha frente al coronavirus. A pesar de que su evaluación excluyó a pacientes vacunados (la mayoría en España actualmente) y que su uso más allá de los 5 días desde el debut de la enfermedad limita su utilidad clínica, parece que su pauta de fácil cumplimiento y su beneficio-riesgo lo pueden convertir en una herramienta importante en los pacientes más susceptibles.
Por su parte, Evusheld® es una asociación de tixagevimab y cilgavimab, dos nuevos anticuerpos monoclonales humanos recombinantes, con una vida media optimizada, producidos a partir de anticuerpos obtenidos de células B de pacientes convalecientes de infección por SARS-CoV-2. Ambos se unen afín, selectiva y simultáneamente a regiones no superpuestas del dominio de unión al receptor (RBD) de la proteína S: impiden la unión del virus al receptor ECA2 en las células humanas y bloquean la infección. El medicamento ha sido autorizado en dos inyecciones intramusculares independientes para la profilaxis de la COVID-19 previa a la exposición al SARS-CoV-2 en adultos y adolescentes a partir de 12 años que pesen ≥ 40 kg.
Su autorización se sustentó en un estudio de fase 3 aún en marcha (N= 5.197), en que el tratamiento en adultos sin COVID-19 ni antecedentes de ella, no vacunados pero con alto riesgo de infección por circunstancias personales o profesionales, o bien con riesgo elevado de respuesta inmune inadecuada a las vacunas (por contraindicación, edad avanzada, inmunodepresión o comorbilidades), demostró una reducción del riesgo de COVID-19 sintomática en un 83% frente a placebo, tras más de 6 meses desde la inyección. No habiéndose registrado ningún caso de enfermedad grave o muerte en el brazo experimental, la eficacia fue independiente de factores como la edad o susceptibilidad de mayor gravedad. Además, con una incidencia de eventos adversos –casi todos leves-moderados– similar a placebo, el medicamento mostró un buen perfil de seguridad en que destacan reacciones locales en la zona de inyección y de hipersensibilidad. Si bien persisten incertidumbres sobre su efectividad frente a la variante ómicron, son los primeros monoclonales anti-proteína S disponibles en España y pueden ser de utilidad en la inmunización pasiva de grupos de riesgo.
INFECCIÓN DE PIEL Y TEJIDOS BLANDOS
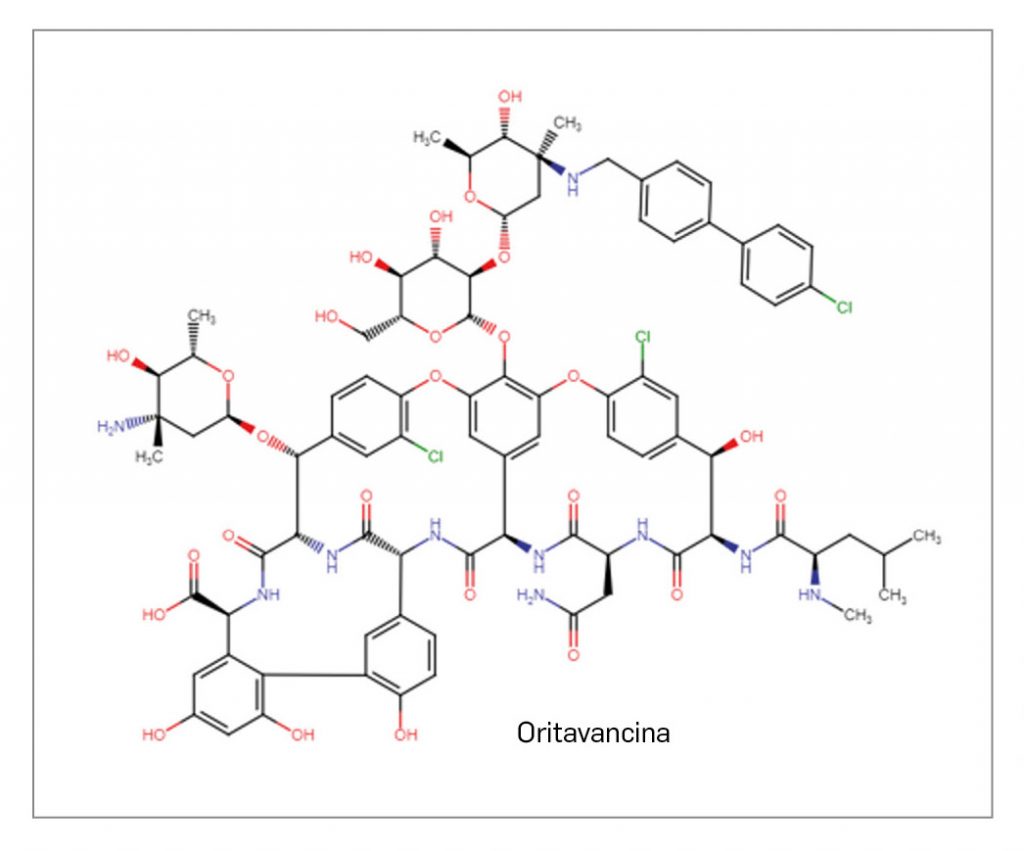
ORITAVANCINA
▼TENKASI (Menarini) PAM 458
Oritavancina es un nuevo lipoglicopéptido que ejerce su efecto antibacteriano por un triple mecanismo sobre la síntesis y reparación de la pared bacteriana (inhibe la transglicosilación y la transpeptidación e interrumpe la integridad de la membrana bacteriana), provocando un aumento de la permeabilidad en dicha pared que permite el contacto directo entre citoplasma bacteriano y el entorno químico externo. Ello resulta en una muerte rápida de las bacterias, que es la base de la autorización del medicamento en el tratamiento, en dosis única intravenosa y según las directrices oficiales del uso de antibacterianos, de pacientes adultos con infecciones agudas de la piel y tejidos blandos de la piel (IPTBs). Presenta un espectro de actividad –limitado– similar a otros glicopéptidos aprobados con igual indicación: microbicida frente a las bacterias grampositivas más frecuentemente implicadas en IPTBs, como Staphylococcus aureus (incluidas las cepas resistentes a meticilina, SARM) y los estreptococos betahemolíticos (como Streptococcus pyogenes) o del grupo viridans.
Su aprobación se sustentó en dos amplios ensayos de fase 3, de adecuado diseño, que probaron la no inferioridad de oritavancina frente a vancomicina (fármaco de elección muy usado frente a SARM) en términos de la tasa de curación clínica tras 7-14 días desde el fin del tratamiento: 80-83% con oritavancina y 80-81% con vancomicina; en pacientes tratados durante más de una semana, se alcanzó una tasa del 91-93% y 89-95%, respectivamente. No se puede concluir respecto a su eficacia frente a infecciones distintas a celulitis, abscesos e infecciones de heridas, en pacientes con septicemia, ni frente a estafilococos con sensibilidad reducida a vancomicina. Tampoco se han realizado comparaciones directas con otros antibióticos, pero los estudios comparativos indirectos disponibles –de robustez limitada– sugieren que oritavancina y dalvabancina, con un mejor balance beneficio-riesgo que telavancina, pueden considerarse como alternativas interesantes y similares a otros antibióticos usados en terapia empírica estándar cuando hay sospecha de SARM, incluido vancomicina.
Se trata de un fármaco relativamente bien tolerado, con un perfil de seguridad comparable al de otros glicopéptidos. La mayoría de las reacciones adversas al fármaco, más frecuentes en mujeres y en pacientes de > 75 años de edad, son leves-moderadas en severidad y no suponen interrupciones del tratamiento (< 4%). Por su frecuencia, destacan las reacciones de hipersensibilidad (12%, como prurito y urticaria), las náuseas (10%), el dolor de cabeza (7%), los vómitos (5%) y los eventos adversos hepáticos (5%); la celulitis es la reacción adversa grave más reportada (1%).
En resumen, oritavancina representará una alternativa más a otras opciones de antibioterapia ambulatoria (linezolid, tedizolid y dalvabancina) y por vía parenteral (vancomicina, linezolid, tedizolid, daptomicina) previamente disponibles. Su elevada vida media (200-300 h) puede ser una ventaja de cara a la adherencia terapéutica (dosis única), por ejemplo, frente a vancomicina o teicoplanina, pero a la vez ser un inconveniente ante problemas de seguridad o cuando se busca el desescalado. Por tanto, en el tratamiento empírico de IPTBs parece más conveniente usar otras alternativas que puedan retirarse fácilmente cuando se conozca el agente causal. A pesar de no suponer ninguna mejora clínica sustancial en su indicación, la incorporación de nuevos agentes antibacterianos con actividad frente a microbios multirresistentes es siempre una buena noticia para la sociedad.
Agentes antineoplásicos e inmunomoduladores
VARIOS TIPOS DE TUMORES SÓLIDOS
CEMIPLIMAB
▼LIBTAYO® (Regeneron Ireland) PAM 455
Cemiplimab es un nuevo anticuerpo monoclonal dirigido frente al receptor de muerte celular programada PD-1: bloquea su interacción con los ligandos PD-L1 y PD-L2 expresados en células tumorales y/u otras del microentorno tumoral, de modo que potencia las respuestas antitumorales de células T. El medicamento ha recibido la autorización condicional para su uso en monoterapia por vía intravenosa para el tratamiento de pacientes adultos con: carcinoma cutáneo de células escamosas (CCE) metastásico o localmente avanzado no candidato para cirugía o radiación curativas; carcinoma basocelular (CBC) localmente avanzado o metastásico que ha progresado tras el uso de un inhibidor de la vía de señalización Hedgehog o en pacientes intolerantes a estos fármacos; y en primera línea de un carcinoma pulmonar no microcítico (CPNM) localmente avanzado (no candidato a quimiorradiación definitiva) o metastásico que exprese PD-L1 ≥ 50% y sin mutaciones en EGFR, ALK o ROS1.
En un estudio de fase 2 abierto y de un solo brazo, cemiplimab indujo, tras una mediana de seguimiento de 8 meses, una tasa de respuesta objetiva del 41% en pacientes con CCE metastásico, tratados o no previamente. Esto se correlacionó con una mediana de supervivencia libre de progresión de 10,4 meses y una tasa de supervivencia global a los 12 meses del 76%. Su aprobación en CBC avanzado/metastásico se basó en los datos derivados de otro estudio de fase 2 con pacientes en recaída o refractariedad tras el uso de inhibidores de la vía Hedgehog o intolerantes a dichos fármacos: el nuevo tratamiento se asoció con una TRO del 29-32% tras seguimientos de entre 9 y 16 meses, si bien la mayoría eran respuestas parciales (25-26%). A la espera de resultados a largo plazo, no se puede concluir aún sobre el mantenimiento de la respuesta antitumoral y un aumento de supervivencia en pacientes con estos dos tipos de tumores cutáneos no melanoma. Su indicación en primera línea del CPNM avanzado o metastásico sin mutaciones oncogénicas (EGFR, ALK o ROS1) ha sido la mejor contrastada: un estudio pivotal de fase 3 con pacientes que sobreexpresan PD-L1 (≥ 50%) y no son candidatos a quimiorradiación probó que cemiplimab prolonga la mediana de supervivencia en casi 8 meses respecto a regímenes dobles de quimioterapia basada en platino (22,1 vs. 14,3), reduciendo en un 32% el riesgo de muerte, si bien la TRO se mantenía en niveles moderados (37% vs. 21%), con una duración inferior a 2 años.
En general, la eficacia clínica de cemiplimab es consistente en los distintos subgrupos de pacientes, independiente de factores como la edad, el estado de expresión de PD-L1 y los tratamientos previos. Por otra parte, su seguridad está en línea con lo conocido para otros anti-PD-1: tiene un perfil toxicológico importante pero manejable clínicamente, con una alta incidencia de eventos adversos relacionados con el tratamiento (30-37% graves) que a menudo conlleva interrupciones (8%). Por su frecuencia y severidad destacan reacciones adversas como dolor musculoesquelético, fatiga, trastornos gastrointestinales y cutáneos, anemia e infecciones del tracto respiratorio. Además, sobresale la posibilidad de reacciones adversas inmunomediadas, pero también se carece de datos a largo plazo.
En definitiva, sin incorporar ninguna novedad en términos de mecanismo de acción (compartido con pembrolizumab y nivolumab), cemiplimab se incorpora como una nueva opción de inmunoterapia similar a otras ya disponibles para tres tipos de tumores sólidos avanzados/metastásicos, en pacientes con escasas opciones de tratamiento para los que sigue existiendo una necesidad de optimizar la farmacoterapia. Sea como fuere, no va a suponer una cura, sino que constituye un tratamiento sintomático paliativo dirigido a mejorar la calidad de vida y aumentar la supervivencia (no asegurado en los tumores cutáneos). Puesto que se recomienda su uso mantenido hasta toxicidad inaceptable o progresión de la patología, persisten incertidumbres en las limitaciones de la evidencia y se requieren más datos a largo plazo para extraer conclusiones sólidas.
ESCLEROSIS MÚLTIPLE
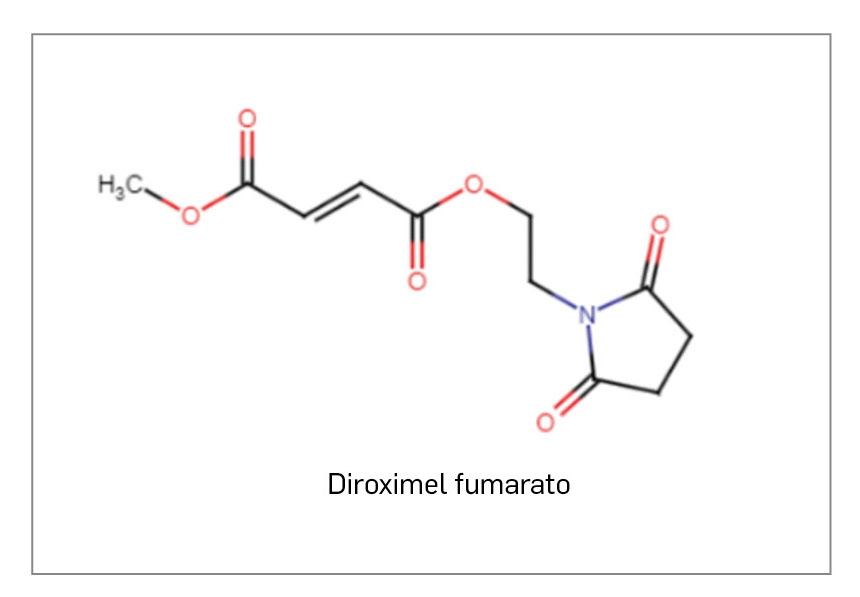
DIROXIMEL FUMARATO
▼VUMERITY® (Biogen Spain) PAM 456
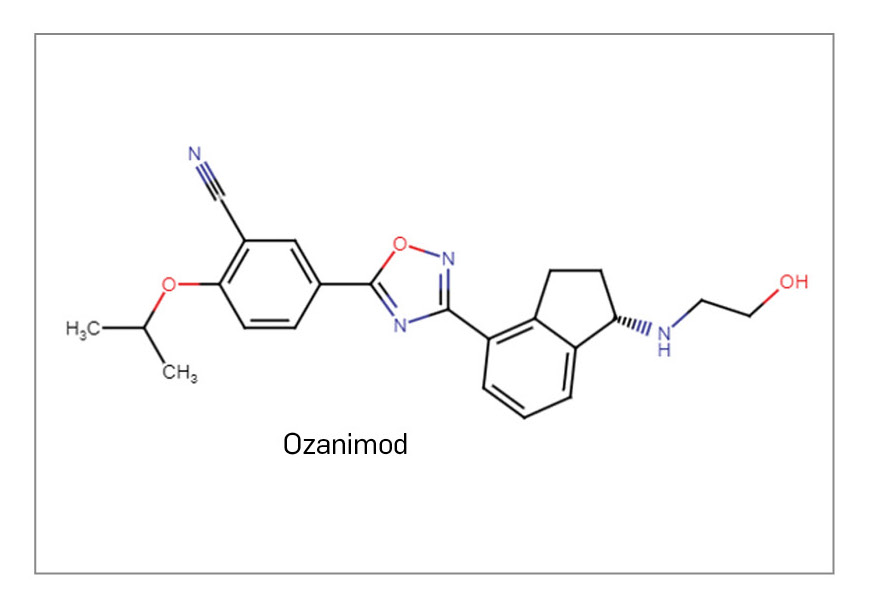
OZANIMOD
▼ZEPOSIA® (Bristol-Myers Squibb) PAM 456
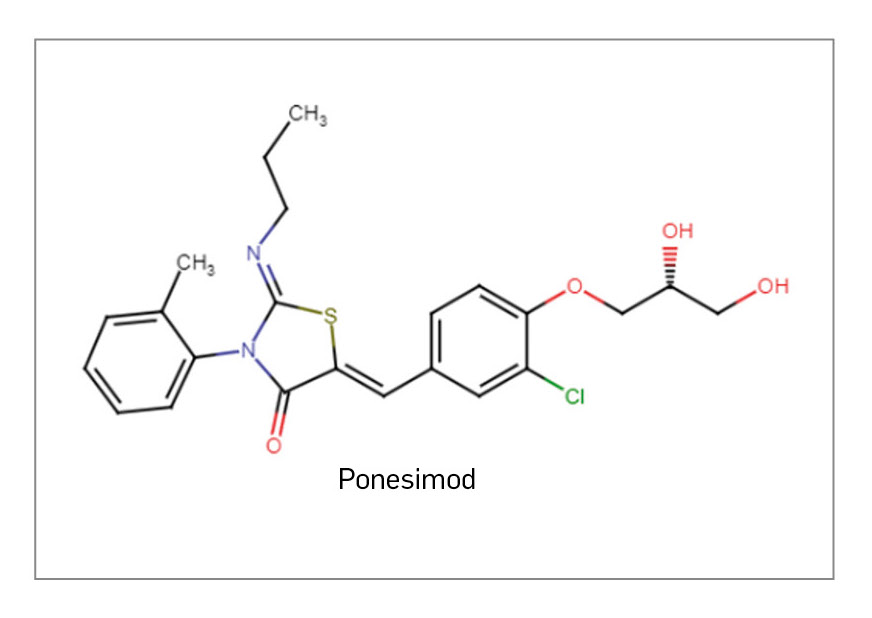
PONESIMOD
▼PONVORY® (Janssen-Cilag) PAM 456
En el número 456 de PAM se analizaron las evidencias disponibles para tres nuevos fármacos activos por vía oral aprobados frente a distintas formas clínicas de la esclerosis múltiple (EM), que no implican a priori ninguna mejora sustancial en relación con el arsenal farmacoterapéutico previamente disponible.
El primero de ellos, diroximel fumarato, es, igual que el ya disponible dimetil fumarato, un profármaco que ejerce sus acciones farmacológicas a través de su principal metabolito activo, el monometil fumarato. No se conocen por completo sus efectos farmacodinámicos, pero parecen mediarse al menos en parte por la activación de la vía de trascripción génica del factor nuclear Nrf2: regula al alza la expresión de los genes antioxidantes y la protección frente al daño por estrés oxidativo, reduciendo la migración de los leucocitos por el endotelio vascular y minimizando el daño axonal y de la mielina de las neuronas. Esto, unido a sus propiedades antiinflamatorias e inmunomoduladoras (reduce la activación de células inmunitarias, el recuento linfocitario y la liberación de citocinas proinflamatorias), subyace tras la autorización del medicamento para el tratamiento de adultos con EM remitente-recurrente (EMRR). Si se extrapolan las consideraciones clínicas aceptadas para dimetil fumarato, el nuevo fármaco se posicionará como una alternativa por vía oral a otros FAMEs disponibles en primera línea de la EMRR (por ejemplo, glatirámero o teriflunomida), de elección en casos sin evolución rápida o alta agresividad de la enfermedad.
También se han comercializado por primera vez otros dos nuevos fármacos activos por vía oral, ozanimod y ponesimod, que comparten mecanismo de acción y se unen al grupo farmacológico de moduladores de receptores de esfingosina, inaugurado por fingolimod y al que ya se había incorporado siponimod en 2021. Al unirse con alta afinidad al receptor S1P1 en los linfocitos, actúan como antagonistas funcionales y previenen sus efectos biológicos: inducen así un “secuestro” de los linfocitos en los ganglios linfáticos, minimizan su infiltración patológica al SNC, y reducen el riesgo de inflamación y lesiones en el tejido nervioso en los pacientes. Ozanimod se ha aprobado para su uso en pacientes adultos con EMRR y enfermedad activa, definida por características clínicas o estudios por imágenes, mientras que ponesimod se ha autorizado para el tratamiento de adultos con cualquier forma de EM en recaída, abarcando la EMRR pero también la EM secundariamente progresiva con brotes superpuestos.
Habiéndose evaluado en ensayos pivotales controlados y de adecuado diseño, han probado su superioridad sobre los comparadores interferón beta y teriflunomida, respectivamente, en términos de reducción de la tasa anualizada de brotes y de variables de resonancia magnética, pero no parecen atenuar en mayor medida la progresión de la discapacidad. Así, pese a ciertas limitaciones de la evidencia, sus IPT establecen que ozanimod y ponesimod pueden considerarse como opciones de tratamiento en primera línea de sus indicaciones, alternativas a sus respectivos comparadores. Serán fármacos interesantes en pacientes con formas activas de EM y factores de mal pronóstico de carácter clínico o radiológico, pero aún se requieren datos a largo plazo (> 2 años) para una mejor caracterización de su balance beneficio-riesgo, necesaria en el contexto de una enfermedad crónica y progresivamente debilitante.
MIELOMA MÚLTIPLE
ISATUXIMAB
▼SARCLISA® (Sanofi Aventis) PAM 453
Isatuximab es un nuevo anticuerpo monoclonal que se une específicamente al receptor CD38, el cual se expresa uniforme e intensamente por las células del mieloma múltiple (MM). Esa unión media la activación de diversos mecanismos citotóxicos e inmunitarios que acaban con la lisis o apoptosis de las células mielomatosas y permiten controlar el crecimiento tumoral. El medicamento ha sido autorizado, en combinación con pomalidomida y dexametasona, para el tratamiento de pacientes adultos con MM resistente al tratamiento o recidivante que han recibido ≥ 2 tratamientos previos, incluyendo lenalidomida y un inhibidor del proteasoma y han mostrado progresión de la enfermedad tras el último tratamiento; y en combinación con carfilzomib y dexametasona para el tratamiento de pacientes adultos con MM que han recibido al menos un tratamiento previo.
Su aprobación se ha sustentado en dos ensayos clínicos de diseño adecuado, que han confirmado su favorable balance beneficio-riesgo. El primero demostró la superioridad de la adición de isatuximab a la combinación de pomalidomida y dexametasona, en comparación con el solo uso de estos dos fármacos como control activo, en pacientes con MM en progresión tras haber recibido ≥ 2 líneas previas, incluyendo lenalidomida y un inhibidor de proteasoma: prolongó la mediana de SLP en unos 5 meses (11,5 vs. 6,5 meses), reduciendo el riesgo de progresión de la enfermedad o muerte en un 40%. También mejoró la tasa de respuesta antitumoral (60% vs. 35%) y su profundidad, resultando en una tendencia favorable en SG –prolongación de la mediana en casi 7 meses (24,6 vs. 17,7)– que aún debe confirmarse con los análisis finales. El segundo estudio evidenció que la asociación del nuevo fármaco a la combinación de carfilzomib y dexametasona mejora los resultados de la doble terapia con estos dos últimos en pacientes con MM en recaída que habían recibido entre 1 y 3 líneas previas de tratamiento (excluyendo carfilzomib y daratumumab): prologó notablemente la SLP (mediana no alcanzada en el brazo experimental vs. 19,1 meses en el control), reduciendo en un 47% el riesgo de progresión o muerte. Sin datos maduros de SG, la tasa y profundidad de la respuesta también fueron mayores con isatuximab (muy buena respuesta parcial o mejor en 73% vs. 56%). La eficacia del nuevo fármaco se reveló rápida, duradera y consistente en los distintos subgrupos de pacientes evaluados, con independencia de la refractariedad a fármacos concretos.
No obstante, su perfil toxicológico es importante y se asocia a una alta frecuencia de reacciones adversas, graves en casi 2 de cada 3 pacientes. Los eventos adversos de mayor incidencia, en su mayoría manejables en clínica, serían esperables por tratarse de una proteína inmunomoduladora y a la vista de la seguridad de los fármacos con que se combina. Sobresalen por su frecuencia (≥ 20%) las infecciones del tracto respiratorio superior e inferior (incluidas bronquitis y neumonía), citopenias (sobre todo, neutropenia), reacciones a la perfusión, alteraciones del tracto gastrointestinal (náuseas y diarrea) y otras inespecíficas, como fatiga o insomnio. Por su gravedad, las infecciones y, en particular, la neumonía, parecen constituir el mayor riesgo asociado al fármaco.
Isatuximab no aporta ninguna novedad en cuanto a mecanismo de acción: comparte vía terapéutica con el ya comercializado daratumumab. Las comparaciones indirectas disponibles entre ambos sugieren que no supera las mejoras incorporadas por daratumumab, el cual tiene un espectro de indicaciones más amplio (incluso en 1ª línea) y se considera de elección en esquemas de rescate. En definitiva, el nuevo fármaco representa una alternativa más de triple terapia a partir de la 2ª línea de tratamiento, y su uso puede ser especialmente relevante en pacientes refractarios a lenalidomida; en ese contexto, se suma a otros fármacos biológicos previamente disponibles (por ejemplo, daratumumab o elotuzumab) sin aportar ningún elemento de innovación terapéutica disruptiva. Además, la futura disponibilidad de las prometedoras terapias CAR-T (idecabtagén vicleucel) hace previsible un cambio en los enfoques de tratamiento.
CÁNCER DE MAMA
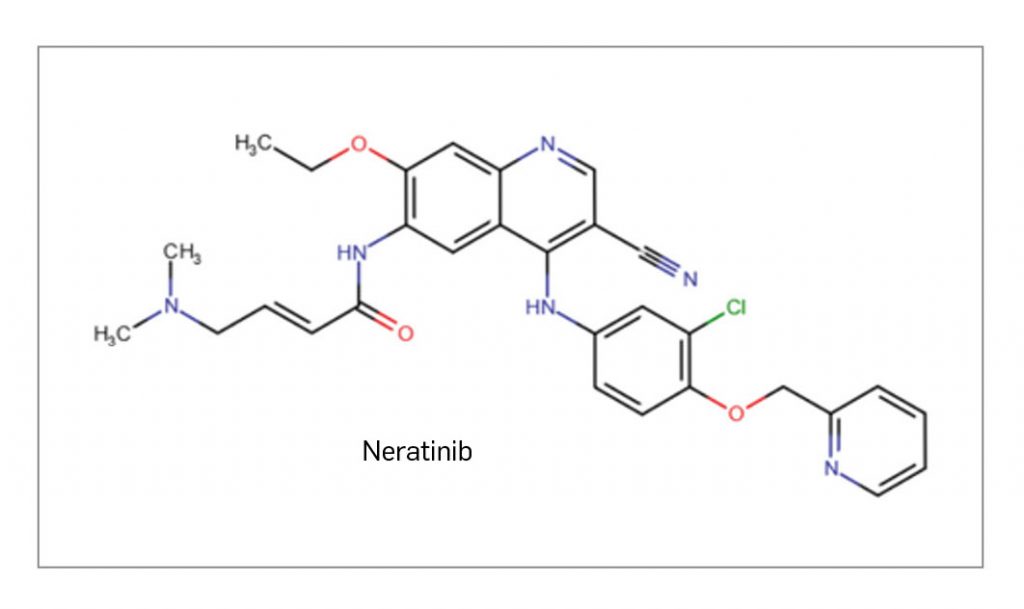
NERATINIB
▼NERLYNX® (Pierre fabre) PAM 457
Neratinib es un nuevo inhibidor selectivo e irreversible la tirosina cinasa del homólogo del oncogén viral de la leucemia paneritroblástica (ErbB). Su acción inhibitoria impide que se desencadene la cascada de señalización intracelular por segundos mensajeros mediante una unión covalente de alta afinidad al sitio de unión del ATP de EGFR (o HER1), HER2 y HER4, o sus heterodímeros activos con HER3. El resultado es una inhibición sostenida de estas vías promotoras del crecimiento en los tumores de mama con amplificación o sobreexpresión de HER2 o mutaciones del mismo, bloqueando la progresión tumoral. El medicamento ha sido autorizado por vía oral en el tratamiento adyuvante extendido de cáncer de mama en estadio inicial con receptor hormonal positivo y sobreexpresión/amplificación de HER2, que hayan finalizado el tratamiento de trastuzumab hace < 1 año.
En el ensayo pivotal aleatorizado y controlado por placebo (n= 2.840), el tratamiento con neratinib se prolongó durante 1 año. Tras los 2 primeros años de seguimiento desde el inicio del estudio se observó una mejora estadísticamente significativa de la supervivencia libre de enfermedad invasiva (SLEi) en el subgrupo de pacientes con tumores positivos a receptores hormonales (RH+), que fue el subgrupo de pacientes que más se benefició, pese a que ese beneficio clínico sigue siendo modesto: la tasa de SLEi fue del 95,3% en el brazo de neratinib y del 90,8% en el brazo placebo (HR: 0,49). Los resultados finales de eficacia tras 5 años de seguimiento confirman la mejora en la SLEi, que en el subgrupo de pacientes RH+ tratadas con neratinib alcanza una tasa del 90,8% frente al 85,7% con placebo, reduciendo a la mitad el riesgo de invasión de la patología (HR: 0,5). Sin embargo, no hubo diferencias estadísticamente significativas en la supervivencia global entre ambas intervenciones a los 8 años de seguimiento. Además, respecto al perfil de seguridad, casi todas las pacientes notifican algún evento con su uso (99% con neratinib vs. 88% con placebo), estando fundamentalmente relacionados con el aparato digestivo. La reacción adversa más frecuente fue la diarrea (95% vs. 35% con placebo) y destaca, además, como el evento adverso de grado ≥ 3 más común (40% vs. 2%), siendo motivo de retirada en una proporción importante de casos (17%); no obstante, parece manejable con el uso de loperamida o ajustes posológicos.
Hasta hace poco, las opciones de tratamiento en adyuvancia en pacientes con cáncer de mama precoz positivo para HER2 y receptores hormonales han sido principalmente las combinaciones de terapia endocrina con quimioterapia y trastuzumab en uso secuencial o concomitante. También se han añadido agentes más recientes, como pertuzumab en pacientes con alto riesgo de recaída, o trastuzumab emtansina en monoterapia en pacientes con enfermedad residual invasiva en mama o ganglios linfáticos tras adyuvancia basada en taxanos y terapia anti-HER2, que representan a día de hoy la mayoría de las pacientes con cáncer de mama precoz HER2+ que han sido tratadas, y a ellas no se pueden extrapolar los datos clínicos disponibles para neratinib, que va dirigido a una fase posterior al uso en adyuvancia de trastuzumab (o quimioterapia + pertuzumab o trastuzumab emtansina). En este contexto, neratinib no ha demostrado cubrir la necesidad terapéutica de mejorar los resultados en supervivencia respecto al solo uso del tratamiento adyuvante con quimioterapia y trastuzumab, por lo que no parece modificar sustancialmente la terapéutica estándar ni incorporar un mecanismo de acción innovador (muy similar al de ya disponible lapatinib).
SACITUZUMAB GOVITECÁN
▼TRODELVY (Gilead) PAM 460
TRASTUZUMAB DERUXTECÁN
▼ENHERTU (Daiichi Sankyo) PAM 460
Se recomienda consultar el artículo monográfico sobre sacituzumab govitecán y trastuzumab deruxtecán que se publica en este mismo número.
HEMOGLOBINURIA PAROXÍSTICA NOCTURNA Y SÍNDROME HEMOLÍTICO URÉMICO ATÍPICO
RAVULIZUMAB
▼ULTOMIRIS® (Alexion Pharma) PAM 456
Se trata de un nuevo anticuerpo monoclonal que se une selectivamente a la proteína C5 del complemento e inhibe de forma inmediata y completa su escisión en C5a y C5b: impide la formación del complejo terminal de ataque a membrana y actúa como un inmunosupresor específico limitando la opsonización de microorganismos y la eliminación de inmunocomplejos. Dado que también bloquea la sobreactivación patológica del complemento que lisaría los eritrocitos sanos, el medicamento ha sido autorizado para el tratamiento por vía intravenosa de pacientes adultos y pediátricos de ≥ 10 kg con hemoglobinuria paroxística nocturna (HPN), tanto si tienen hemólisis con manifestaciones indicativas de alta actividad de la enfermedad como si han sido pretratados con éxito con eculizumab los últimos 6 meses; igualmente, para el tratamiento de pacientes de ≥ 10 kg con síndrome hemolítico urémico atípico (SHUa) naïve a inhibidores del complemento o adecuadamente tratados con eculizumab durante ≥ 3 meses.
Diseñado a partir de eculizumab (con el que comparte mecanismo de acción), el nuevo fármaco ha sido evaluado para su uso en HPN en dos estudios pivotales de adecuado diseño (aleatorizados, abiertos y con comparador activo), de 6 meses de duración, que han confirmado su no inferioridad frente a eculizumab en variables relativas a la hemólisis, la anemia y la calidad de vida. Así, hasta un 73% de los pacientes naïve a inhibidores del complemento no necesitó transfusiones plasmáticas (vs. 66% con eculizumab), y entre los pacientes controlados previamente con eculizumab, también una gran mayoría se mantuvo independiente de transfusiones (88% vs. 83%). Por otro lado, su aprobación en SHUa se sustentó en dos ensayos pivotales, también abiertos, pero de un solo brazo y con menor número de pacientes. En adultos naïve, un tratamiento de 6 meses proporciona una tasa de respuesta clínica completa del 54% según parámetros hematológicos y de función renal. El estudio pediátrico reflejó una respuesta clínica incluso mayor (78%) en niños naïve con SHUa; además, probó que quienes cambian desde eculizumab mantienen estables los parámetros de la microangiopatía trombótica. Ravulizumab redujo drásticamente la necesidad de diálisis en aquellos pacientes que antes del tratamiento la requerían. En líneas generales, su eficacia es consistente en los distintos subgrupos y parece mantenerse en el tiempo al menos durante periodos superiores a 1 año.
En términos de seguridad, es un fármaco con un perfil toxicológico benigno, caracterizado mayoritariamente por efectos adversos leves-moderados, que no implican gran número de interrupciones del tratamiento. Las reacciones adversas descritas con mayor frecuencia (hasta 25-30%) son: cefalea, alteraciones gastrointestinales (diarrea, náuseas y vómitos) e infecciones del tracto respiratorio superior (faringitis). Se debe prestar atención al riesgo de infección o sepsis meningocócica, descrito para eculizumab y manejable mediante profilaxis. Aún se requiere una mejor caracterización de la seguridad a largo plazo.
En definitiva, se posiciona como una alternativa de tratamiento a eculizumab en las dos indicaciones aprobadas, con un balance beneficio-riesgo similar, sobre todo en pacientes con requerimientos transfusionales altos, alto grado de anemia hemolítica intravascular y/o episodios agudos de debut de la patología, no debiéndose usar como rescate en casos refractarios a eculizumab. Pese a que no aporta una innovación disruptiva frente a eculizumab, la principal ventaja del nuevo fármaco es la referente a su pauta de administración más espaciada (cada 8 meses vs. 2 meses).
ACONDICIONAMIENTO PRE-TRASPLANTE DE PROGENITORES HEMATOPOYÉTICOS
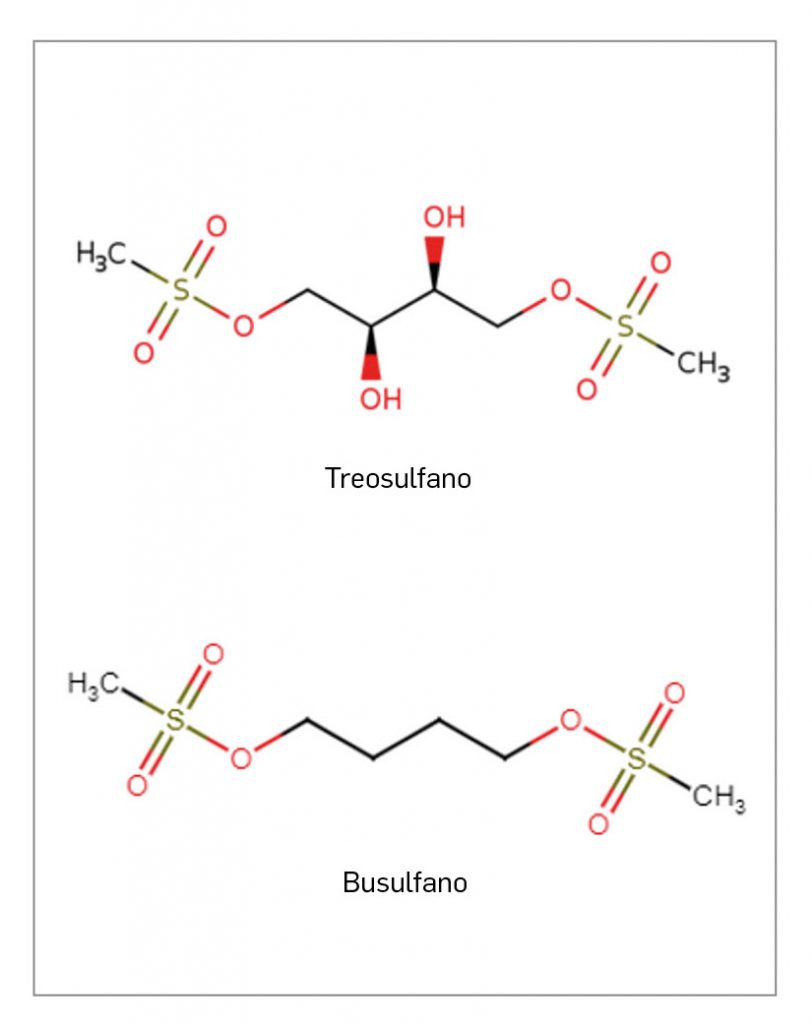
TREOSULFANO
▼TRECONDI® (Gebro Pharma) PAM 457
Treosulfano es un profármaco de un alquilante bifuncional con actividad citotóxica para células progenitoras hematopoyéticas. Su actividad se debe a la conversión espontánea en condiciones fisiológicas a un intermediario monoepóxido y, finalmente, al derivado L-diepoxibutano. Los epóxidos formados alquilan centros nucleofílicos del ADN y son capaces de inducir entrecruzamientos de ADN que se consideran responsables de los efectos antineoplásicos y de la disminución de las células progenitoras. Se encuentra autorizado en España como medicamento huérfano y está indicado como parte de un tratamiento de acondicionamiento previo a trasplante alogénico de progenitores hematopoyéticos en combinación con fludarabina, tanto en pacientes adultos con neoplasias malignas y enfermedades benignas como en pacientes pediátricos mayores de un mes con neoplasias malignas.
Se llevó a cabo un ensayo pivotal aleatorizado, de fase 3, de grupos paralelos y con comparador activo en adultos (N= 460). El análisis de la supervivencia libre de enfermedad (SLE) arrojó un resultado positivo estadísticamente significativo para treosulfano en comparación con busulfano (HR: 0,65), demostrando así la no inferioridad. También se observó una mejora en el análisis de las variables secundarias, como la supervivencia global (HR= 0,61) –la variable más robusta en oncología– o en la mortalidad relacionada con el tratamiento (HR= 0,54). Los resultados de eficacia a 3 años mostraron la superioridad de treosulfano frente a busulfano en cuanto al objetivo de SLE (HR= 0,64; p= 0,0006). Para la población pediátrica se ha desarrollado un estudio específico (N= 70) abierto, no controlado y de fase 2, en el que la supervivencia global a los 12 meses fue del 91,4%. Durante este periodo de seguimiento se produjeron 7 fallecimientos, confirmándose que la MRT a los 12 meses fue de solo el 2,9%. Los eventos adversos más frecuentemente observados fueron similares en adultos y población pediátrica, principalmente eventos gastrointestinales como mucositis oral, náuseas o vómitos y también un aumento en las infecciones, elemento que constituyó la principal reacción adversa grave. No obstante, la frecuencia y gravedad observadas en los eventos adversos fue superior en la población adulta.
Dado el perfil más favorable de seguridad respecto al acondicionamiento con busulfano, así como una eficacia en la supervivencia libre de enfermedad que se ha demostrado no inferior, se puede considerar que treosulfano, a pesar de tratarse de una modificación químicamente sencilla de busulfano y carecer de un mecanismo de acción novedoso, aporta una mejora notable al pronóstico de ciertas neoplasias hematológicas para las que el TPH representa la única opción curativa y en las que, hasta la fecha, las posibilidades de recurrir a este tratamiento se han visto seriamente limitadas por su elevada toxicidad. Así, el nuevo fármaco abre una oportunidad para ampliar este tratamiento a un mayor número de pacientes y a otras enfermedades en las que, hasta ahora, el balance beneficio/riesgo se había considerado desfavorable.
MACROGLOBULINEMIA DE WALDESTRÖM
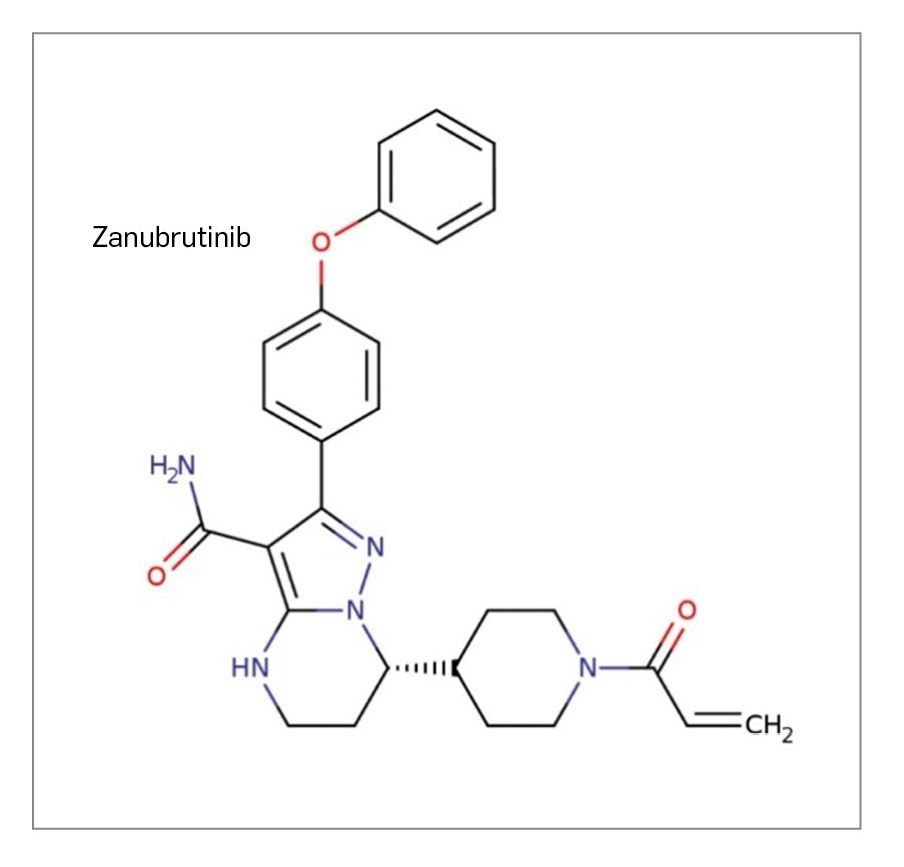
ZANUBRUTINIB
▼BRUKINSA® (Beigene) PAM 459
Zanubrutinib es un nuevo inhibidor de tirosina cinasa de Bruton (BTK) de segunda generación que, como ibrutinib, se une a esta de forma irreversible en el sitio unión del ATP. La BTK es una molécula de señalización del receptor antigénico de linfocitos B y las vías receptoras de citocinas, cuya activación conduce a la fosforilación de la fosfolipasa PLCγ2, activando de este modo factores de transcripción necesarios para la proliferación, la quimiotaxis y la adhesión de las células B. Se comprende, por tanto, que su inhibición por zanubrutinib tenga efectos antiproliferativos en esta estirpe celular. El medicamento ha sido autorizado para su uso en monoterapia para el tratamiento por vía oral de pacientes adultos con macroglobulinemia de Waldenström (MW) que han recibido al menos una terapia previa o como tratamiento de primera línea en pacientes que no son aptos para quimioinmunoterapia.
Su eficacia y seguridad de zanubrutinib se han evaluado adecuadamente en un único estudio pivotal con pacientes con MW, de fase 3, aleatorizado, abierto, controlado por ibrutinib y multicéntrico. El estudio se compuso de dos cohortes, una compuesta por pacientes con mutación en el gen MYD88 (n= 201) que fueron asignados (1:1) a zanubrutinib o ibrutinib, y una segunda cohorte compuesta de un único brazo de pacientes sin mutación en MYD88 o estado mutacional desconocido (n= 28). Ningún paciente alcanzó la respuesta completa en ninguno de los brazos de tratamiento. En la primera cohorte, un número mayor de pacientes consiguió una muy buena respuesta parcial con zanubrutinib que con ibrutinib (28% vs. 19%), tanto entre pacientes naïve como entre los que estaban en recaída/refractariedad tras un tratamiento previo, pero la diferencia no resultó estadísticamente significativa (p= 0,1160). Si bien el resto de variables se consideraron descriptivas, es destacable que el tiempo hasta alcanzar MBRP fue menor con zanubrutinib que con ibrutinib (5,5 vs. 22,1 meses) en pacientes sin tratamiento previo, pero esa diferencia se redujo considerablemente en pacientes en recaída o refractariedad (6,6 vs. 7,9 meses), manteniendo zanubrutinib una ligera ventaja. No se vieron diferencias significativas en términos de supervivencia por no haberse alcanzado las medianas de SLP ni de SG.
Por otra parte, el perfil de seguridad de zanubrutinib parece similar e incluso ligeramente más favorable que el de ibrutinib, con menor incidencia de algunos de sus eventos adversos característicos (fibrilación auricular, diarrea e hipertensión). Se caracteriza por una baja proporción de pacientes que suspenden el tratamiento por eventos adversos (4% vs. 9% con ibrutinib). La reacción adversa más frecuente con el uso de zanubrutinib fue la neutropenia (24% vs. 12% con ibrutinib), pero no se observaron diferencias en cuanto a la incidencia de infecciones entre los brazos de tratamiento.
A pesar de que zanubrutinib no incorpora un mecanismo de acción novedoso, parece que su especificidad por la inhibición de BTK es mayor que la de ibrutinib, lo que puede relacionarse con un perfil de seguridad algo más favorable. El nuevo fármaco se incorpora al arsenal terapéutico de la MW como una opción más cuya utilidad es similar a la del ya aprobado y comercializado ibrutinib, aunque puede representar una alternativa de tratamiento para algunos pacientes en los que ciertas reacciones adversas a éste, como las cardiovasculares, desaconsejen su empleo. No parece que vaya a modificar sustancialmente la terapéutica de la enfermedad.
Sistema musculoesquelético
OSTEOPOROSIS EN MUJERES POSMENOPÁUSICAS
ROMOSOZUMAB
▼EVENITY® (UCB Pharma) PAM 458
Romosozumab es un nuevo anticuerpo monoclonal que se une selectivamente a la esclerostina e impide la función normal de esta proteína (inhibir la proliferación y la función de los osteoblastos), ejerciendo un doble efecto: aumenta la formación de hueso y disminuye la resorción ósea. Por tanto, produce aumentos rápidos en la masa ósea y mejoras en su estructura y resistencia, lo cual ha sido la base para la autorización del medicamento en el tratamiento por vía subcutánea de la osteoporosis grave en mujeres posmenopáusicas con un elevado riesgo de fractura. Se debe instaurar una suplementación diaria de calcio y vitamina D.
En un ensayo pivotal de fase 3, que incluyó a pacientes mayores (media de 74 años de edad) con osteoporosis grave y antecedentes de fracturas por fragilidad (N > 4.000), el tratamiento de 12 meses con el fármaco probó su superioridad frente a alendronato semanal, en ambos casos seguido de otros 12 meses de terapia con alendronato. Se evidenció una reducción significativa de la tasa de fracturas vertebrales: a los 24 meses, redujo el riesgo relativo de fracturas a la mitad (tasa de 4,1% vs. 8,0% con alendronato). El análisis primario, tras un seguimiento de 33 meses, reveló también un descenso en la incidencia de fracturas clínicas (9,7% vs. 13%) y de fracturas de cadera (2,0% vs. 3,2%), asociados a una mejora sustancial de la densidad mineral ósea. Un segundo estudio comparado con placebo en pacientes con osteoporosis más leve (N > 7.000) reflejó que el tratamiento con romosozumab por 12 meses seguido del mismo tiempo con denosumab reduce la incidencia de fracturas vertebrales tras 2 años (0,6% vs. 2,5% con placebo), pero no de fracturas clínicas o de cadera.
En términos de seguridad, su perfil toxicológico se asocia con la incidencia de infecciones, como nasofaringitis (14%), artralgia (12%) o reacciones de hipersensibilidad (7%). No obstante, el signo de seguridad más importante es el aumento relativo (+70%) del riesgo de eventos cardiovasculares graves (ictus e infarto de miocardio) y una mayor mortalidad asociada, lo que contraindica su uso ante antecedentes cardiovasculares y obliga a evaluar de forma individualizada cada caso; pese a todo, se esperarían 2 eventos cardiovasculares graves adicionales –sobre todo, ictus o infarto– por cada 1.000 pacientes tratadas. Estos motivos hicieron que la EMA rechazara inicialmente la solicitud de autorización de este medicamento.
En definitiva, romosozumab inaugura una vía terapéutica en osteoporosis grave y su uso durante un año ha probado cierto beneficio por periodos de hasta 3 años en pacientes con alto riesgo de fractura. El mayor interés de su uso radicaría en su capacidad de reducir la incidencia de fracturas de cadera –las fracturas osteoporóticas de mayor impacto clínico y mortalidad–, pero la magnitud del efecto parece moderada. Por tanto, la dudosa relevancia clínica de los efectos observados, junto a las incertidumbres de la seguridad cardiovascular y de su efecto a largo plazo, ponen en entredicho su grado de innovación. Teniendo en cuenta que en la indicación se dispone de alternativas con un perfil diferente pero mejor conocidas (sobre todo, los bisfosfonatos alendronato, risedronato o zoledronato), que no se han probado ventajas en adherencia terapéutica para el nuevo fármaco, y que tras los 12 meses de tratamiento se requiere continuar con un tratamiento anti-resortivo, no parece que vaya a suponer un cambio de paradigma en el tratamiento de la osteoporosis. No ha sido financiado en España para su uso en el Sistema Nacional de Salud.
Sistema nervioso
EPILEPSIA FOCAL REFRACTARIA
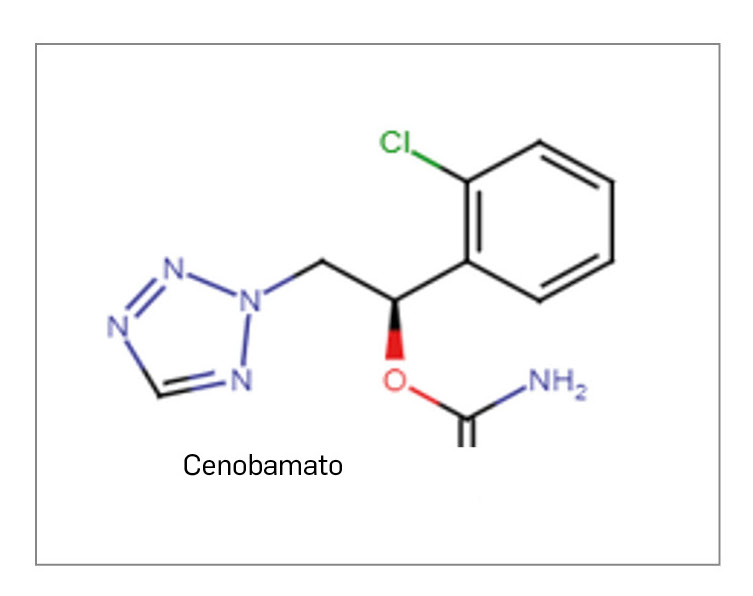
CENOBAMATO
▼ONTOZRY (Angelini Pharma) PAM 459
Cenobamato es un nuevo antiepiléptico cuyo mecanismo de acción no ha sido del todo dilucidado. Se ha postulado que ejerce una acción dual: actúa como modulador alostérico positivo del receptor del canal iónico del GABA (GABAA) y bloquea e inactiva de forma más prolongada los canales de Na+, consiguiendo reducir la excitabilidad neuronal y la descarga repetitiva. El medicamento ha sido autorizado para el tratamiento concomitante por vía oral de las crisis de inicio focal con o sin generalización secundaria en adultos con epilepsia que no han sido controlados de forma adecuada a pesar del tratamiento previo con al menos 2 antiepilépticos.
Los resultados de dos estudios de fase 2 de similar diseño, uno pivotal y otro de soporte, han revelado que la adyuvancia con cenobamato durante un mantenimiento de 6-12 semanas a dosis fija –tras un periodo de titulación previo– se asocia con una alta tasa de pacientes respondedores (50-64%), significativamente superior a placebo (20-27%), y con una marcada reducción en la frecuencia de crisis, también significativa respecto a placebo (de -55% a -63% vs. -22% a -27%). En la fase de extensión del estudio pivotal se probó que su eficacia es consistente en los distintos subgrupos de pacientes e independiente del tratamiento previo y concomitante; además, se mantiene a largo plazo: tras 25-30 meses reduce la frecuencia de crisis en un 76% y hasta un 20% de los pacientes están libres de crisis. Por otra parte, el perfil toxicológico del nuevo fármaco es dependiente de la pauta posológica, similar al de otros antiepilépticos y con una tolerabilidad similar a placebo. Las reacciones adversas más frecuentes (> 10%) se relacionan con el sistema nervioso central –somnolencia, mareo, fatiga y cefalea–, en su mayoría leves-moderadas y de corta duración; entre las graves (14%), destacan la ataxia, los mareos y la somnolencia como principales causas de discontinuación. Una titulación lenta del fármaco desde una dosis inicial baja minimiza el riesgo de DRESS.
Habiéndose solo investigado frente a placebo, no se dispone de comparaciones directas de cenobamato con otros antiepilépticos. Las comparaciones indirectas podrían sugerir que la tasa de pacientes libres de crisis (≈20%) supera la observada en los ensayos con otros fármacos anticrisis usados en epilepsia focal (< 7%), como lacosamida, eslicarbazepina, lamotrigina o los más recientes brivaracetam o perampanel; de forma similar, las altas tasas de retención con cenobamato a largo plazo (cercanas al 60% a los 6 años) serían superiores a las descritas para otros fármacos anticrisis.
En definitiva, los adultos con crisis de inicio focal farmacorresistentes –que no hayan conseguido un control adecuado o no toleren al menos 2 líneas previas (una primera monoterapia y una segunda con otra opción de monoterapia o con una primera adyuvancia)– se podrán beneficiar del uso diario por vía oral de cenobamato como alternativa en una 3ª línea de tratamiento como primera o segunda adyuvancia; muy probablemente, tras el uso de un antiepiléptico de 3ª generación (como brivaracetam, perampanel, eslicarbazepina o lacosamida). Cenobamato no incorpora un mecanismo de acción novedoso en la indicación ni parece suponer un cambio de paradigma en el abordaje de la epilepsia, pero sí va a cubrir una laguna terapéutica por tratarse del primer medicamento específicamente autorizado para el tratamiento concomitante tras el fallo de 2 o más fármacos. Su lenta titulación y el hecho de no estar disponible en formulación intravenosa impiden su empleo en situaciones de urgencias.
TRASTORNO DEPRESIVO MAYOR
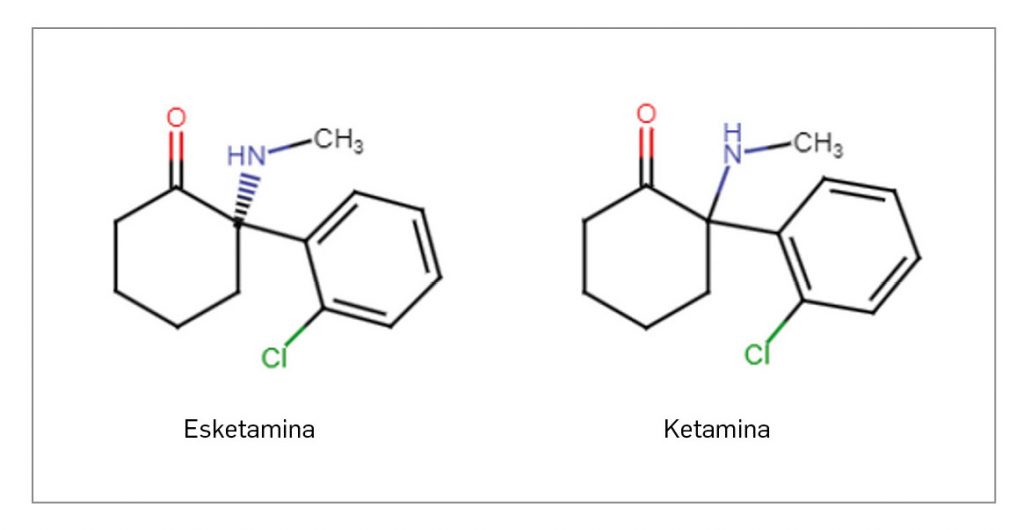
ESKETAMINA
▼SPRAVATO (Janssen-Cilag) PAM 459
Esketamina es un nuevo antidepresivo que, desde un punto de vista químico, es el enantiómero S de la ketamina. Su acción se media por un antagonismo no selectivo y no competitivo del receptor ionotrópico NMDA de glutamato, que produce un aumento transitorio de la liberación de glutamato y una mayor estimulación de su receptor AMPA. Así, puede contribuir al restablecimiento de la función sináptica en las regiones cerebrales implicadas en la clínica de la depresión. En base a ello, el medicamento ha sido autorizado para el tratamiento por vía intranasal, en combinación con un ISRS o IRSN, de adultos con trastorno depresivo mayor (TDM) resistente al tratamiento, que no han respondido al menos a dos tratamientos diferentes con antidepresivos en el episodio depresivo moderado o grave actual. También se indica, coadministrado con antidepresivos orales, en el tratamiento agudo a corto plazo en adultos con un episodio de TDM moderado-grave, para la rápida reducción de los síntomas depresivos que se consideran emergencia psiquiátrica según criterio clínico.
El balance beneficio-riesgo de esketamina –adicionada a un antidepresivo oral de nuevo inicio– se ha evaluado en hasta 5 ensayos clínicos aleatorizados de fase 3 que enrolaron a más de 1.800 pacientes adultos con depresión moderada-grave resistente al tratamiento con al menos dos fármacos antidepresivos. Tres de los estudios fueron a corto plazo (4 semanas) con un diseño similar: doble ciego y controlados con un placebo equivalente. En uno de ellos demostró una eficacia significativa, con una diferencia entre tratamientos de -3,5 puntos en el cambio de la escala MADRS al día 28 (variable principal), lo que se considera clínicamente relevante, pero de efecto modesto; en los otros dos estudios a corto plazo no se alcanzó significación estadística, lo que genera incertidumbre por la inconsistencia de los resultados. Entre los estudios de mayor duración destaca un estudio de prevención de recaídas en el que se probó que la adición de esketamina al tratamiento de referencia y su continuación en el tiempo era superior a placebo en la prolongación del tiempo hasta la recaída, con una reducción de un 51% del riesgo de recaída y un evento prevenido por cada 6 pacientes tratados. Por último, su indicación en urgencia psiquiátrica se basó en dos estudios de fase 3 a corto plazo (4 semanas), donde se verificó una eficacia antidepresiva consistente (diferencia de -3,8 puntos frente a placebo en la escala MADRS) y significativa tras las primeras 24 h desde la administración, mayor incluso en pacientes con intentos de suicidio previo. No obstante, su eficacia a largo plazo en la prevención del suicidio no ha sido establecida.
En términos de seguridad, un amplio porcentaje de pacientes notifica algún evento adverso posiblemente relacionado con el fármaco (58-78% vs. 46-64% con placebo), pero la gran mayoría son leves-moderados y autolimitados. Destacan por su frecuencia reacciones adversas que afectan al sistema nervioso, psiquiátricas y gastrointestinales, tales como mareos, disociación, náuseas, cefalea y somnolencia. También es característica la aparición de hipertensión arterial, que requiere precaución, pero el principal riesgo de esketamina se relaciona con el potencial de abuso y uso ilícito, similar al de ketamina.
En definitiva, el nuevo fármaco incorpora un mecanismo de acción novedoso en el abordaje de la depresión, algo que no ocurría desde hace varias décadas, y constituye el primer tratamiento específicamente aprobado en España para pacientes con depresión resistente al tratamiento. A la vista del beneficio clínico modesto que aporta y las dudas sobre su efecto en la prevención del suicidio, no parece que esketamina suponga un cambio de paradigma en el tratamiento. Además, su dispensación y uso se restringen por ahora a centros sanitarios, con supervisión (durante y después) por profesionales sanitarios, que podría limitar la adherencia al tratamiento.
Órganos de los sentidos
DEGENERACIÓN MACULAR ASOCIADA A LA EDAD
BROLUCIZUMAB
▼BEOVU (Novartis) PAM 454
Se trata de un fragmento de anticuerpo monoclonal humanizado, de cadena única, que se une con alta afinidad a las isoformas del VEGF-A (VEGF165, VEGF110 y VEGF121) e impide su unión a sus receptores VEGFR-1 y VEGFR-2. Así, brolucizumab inhibe la proliferación de células endoteliales, reduciendo la neovascularización patológica y disminuyendo la permeabilidad vascular: en consecuencia, reduce el edema retiniano y mejora la función visual. El medicamento ha sido autorizado para su administración intravítrea en el tratamiento de la degeneración macular asociada a la edad (DMAE) neovascular y de la alteración visual debida al edema macular diabético.
En dos ensayos pivotales de fase 3 y doble ciego ha probado su no inferioridad frente a aflibercept en pacientes con DMAE neovascular: la diferencia media en la mejor agudeza visual corregida a la semana 48 fue similar entre ambos brazos (-0,2 y -0,7 letras), confirmándose una respuesta clínicamente relevante (mejora de ≥ 15 letras) en una proporción de pacientes comparable (29-34% vs. 25-29% con aflibercept). Las variables secundarias, incluidas las referentes a parámetros anatómicos y de calidad de vida, confirman la eficacia de brolucizumab, que se mantiene al menos 2 años. De forma similar, en pacientes con edema macular diabético aleatorizados en otros dos estudios pivotales, se puso de manifiesto el beneficio del tratamiento con del nuevo fármaco sobre la agudeza visual: fue no inferior a aflibercept en términos del cambio medio de la mejor agudeza visual corregida a la semana 52 (diferencia de entre -1,3 y +1,2 letras); tenían una mejora de agudeza visual de ≥ 15 letras el 36-47% de los pacientes tratados (vs. 37-40% con aflibercept), y el efecto sobre la retinopatía diabética en ambos brazos era parejo. Por otra parte, el perfil de seguridad de brolucizumab es globalmente benigno y parecido al de otros anti-VEGF: sobresalen las reacciones adversas asociadas a la propia técnica de administración, en su mayoría leves y transitorias; se notifican casos graves en < 1% de los pacientes. Las reacciones adversas más frecuentes son de tipo ocular, tales como: reducción de la agudeza visual, cataratas o hemorragia conjuntival. La inmunogenicidad del fármaco no parece afectar a su eficacia clínica, pero se asocia con eventos de inflamación ocular, por lo que se debe seguir investigando.
A falta de comparaciones directas con ranibizumab (y bevacizumab), todo apunta a que brolucizumab puede ser similar a los demás agentes anti-VEGF, no incorporando ninguna innovación en cuanto a mecanismo de acción. Sin embargo, la tecnología más avanzada respecto a otros fármacos del grupo permite un distanciamiento en el tiempo entre dosis de hasta 3 meses en pacientes sin actividad de la enfermedad, frente a intervalos que no suelen superar las 6-8 semanas con otros fármacos del grupo. Esa ventaja posológica puede ser el factor más relevante a su favor, con potencial para favorecer la adherencia terapéutica y aliviar la carga asistencial. A priori, podría posicionarse como una alternativa más a aflibercept y ranibizumab (y bevacizumab), pero las incertidumbres sobre la evidencia han conducido a su no financiación por el Ministerio de Sanidad y limitan por ahora su consideración al mismo nivel de preferencia, por lo que no supone una modificación sustancial de la terapéutica estándar. Se debe aún aclarar, por ejemplo, si es eficaz y seguro en pacientes que han fracasado a otros fármacos del grupo (solo se ha evaluado en pacientes naïve) y sus resultados clínicos en la pauta de “tratar y extender” comúnmente usada en la práctica clínica.