Resumen
Cenobamato es un nuevo antiepiléptico cuyo mecanismo de acción no ha sido del todo dilucidado. Se ha postulado que ejerce una acción dual: actúa como modulador alostérico positivo del receptor del canal iónico del GABA (GABAA) y bloquea e inactiva de forma más prolongada los canales de Na+, consiguiendo reducir la excitabilidad neuronal y la descarga repetitiva. El medicamento ha sido autorizado para el tratamiento concomitante por vía oral de las crisis de inicio focal con o sin generalización secundaria en adultos con epilepsia que no han sido controlados de forma adecuada a pesar del tratamiento previo con al menos 2 antiepilépticos.
Los resultados de dos estudios de fase 2 de similar diseño, uno pivotal y otro de soporte, han revelado que la adyuvancia con cenobamato durante un mantenimiento de 6-12 semanas a dosis fija –tras un periodo de titulación previo– se asocia con una alta tasa de pacientes respondedores (50-64%), significativamente superior a placebo (20-27%), y con una marcada reducción en la frecuencia de crisis, también significativa respecto a placebo (de -55% a -63% vs. -22% a -27%). En la fase de extensión del estudio pivotal se probó que su eficacia es consistente en los distintos subgrupos de pacientes e independiente del tratamiento previo y concomitante; además, se mantiene a largo plazo: tras 25-30 meses reduce la frecuencia de crisis en un 76% y hasta un 20% de los pacientes están libres de crisis. Por otra parte, el perfil toxicológico del nuevo fármaco es dependiente de la pauta posológica, similar al de otros antiepilépticos y con una tolerabilidad similar a placebo. Las reacciones adversas más frecuentes (> 10%) se relacionan con el sistema nervioso central –somnolencia, mareo, fatiga y cefalea–, en su mayoría leves-moderadas y de corta duración; entre las graves (14%), destacan la ataxia, los mareos y la somnolencia como principales causas de discontinuación. Una titulación lenta del fármaco desde una dosis inicial baja minimiza el riesgo de DRESS.
Habiéndose solo investigado frente a placebo, no se dispone de comparaciones directas de cenobamato con otros antiepilépticos. Las comparaciones indirectas podrían sugerir que la tasa de pacientes libres de crisis (≈20%) supera la observada en los ensayos con otros fármacos anticrisis usados en epilepsia focal (< 7%), como lacosamida, eslicarbazepina, lamotrigina o los más recientes brivaracetam o perampanel; de forma similar, las altas tasas de retención con cenobamato a largo plazo (cercanas al 60% a los 6 años) serían superiores a las descritas para otros fármacos anticrisis.
En definitiva, los adultos con crisis de inicio focal farmacorresistentes –que no hayan conseguido un control adecuado o no toleren al menos 2 líneas previas (una primera monoterapia y una segunda con otra opción de monoterapia o con una primera adyuvancia)– se podrán beneficiar del uso diario por vía oral de cenobamato como alternativa en una 3ª línea de tratamiento como primera o segunda adyuvancia; muy probablemente, tras el uso de un antiepiléptico de 3ª generación (como brivaracetam, perampanel, eslicarbazepina o lacosamida). Cenobamato no incorpora un mecanismo de acción novedoso en la indicación ni parece suponer un cambio de paradigma en el abordaje de la epilepsia, pero sí va a cubrir una laguna terapéutica por tratarse del primer medicamento específicamente autorizado para el tratamiento concomitante tras el fallo de 2 o más fármacos. Su lenta titulación y el hecho de no estar disponible en formulación intravenosa impiden su empleo en situaciones de urgencias.
Aspectos fisiopatológicos
La epilepsia (palabra derivada del griego epilambanein, que significa “apoderarse de” o “atacar”) es uno de los trastornos neurológicos más comunes del sistema nervioso central (SNC), caracterizado por la presencia y recurrencia de crisis transitorias y autolimitadas derivadas de la función anormal –excesiva– de neuronas de la corteza cerebral. Se define una crisis cerebral como cualquier episodio brusco y transitorio de carácter motor, sensitivo, sensorial y psíquico, consecutivo a una disfunción pasajera, parcial o global del cerebro. Por su parte, una crisis epiléptica sería la “expresión clínica de aquella crisis cerebral que resulta de una descarga neuronal sincrónica de alta frecuencia en una zona del cerebro, que se repetirá a lo largo del tiempo y es el resultado de una afección crónica”.
Desde el año 2005 se ha venido definiendo conceptualmente la epilepsia como un trastorno cerebral caracterizado por una predisposición continuada a la generación de crisis epilépticas, esto es, “una condición caracterizada por la aparición con más de 24 h de separación de dos (o más) crisis epilépticas recurrentes no provocadas por alguna causa inmediatamente identificable”.
La International League Against Epilepsy (ILAE, Liga Internacional contra la Epilepsia) revisó la definición en 2014 para incluir los casos especiales que no responden a ese criterio, y aportó una definición clínica práctica de epilepsia como “una enfermedad cerebral definida por cualquiera de las situaciones siguientes: i) aparición de al menos 2 crisis no provocadas (o reflejas) con una separación de > 24 h; ii) aparición de una crisis no provocada (o refleja) y una probabilidad de que aparezcan más crisis durante los 10 años siguientes similar al riesgo de recurrencia general (≥ 60%) después de 2 crisis no provocadas; o iii) diagnóstico de un síndrome epiléptico1” (Fisher et al., 2014).
La epilepsia afecta a alrededor de 70 millones de personas en todo el mundo, y se erige como una de las patologías neurológicas más prevalentes. Según los datos divulgados del estudio Epiberia, su tasa de prevalencia en la población española mayor de 18 años es de casi 15 casos por cada 1.000 habitantes, o sea, actualmente algo más de 700.000 personas viven con la enfermedad en nuestro país, sin haberse detectado diferencias significativas en cuanto a regiones. La tasa de prevalencia de epilepsia activa –pacientes con sintomatología que requieren intervención médica y recursos sanitarios– ronda los 6 casos/1.000 habitantes, lo cual supone algo más de 260.000 pacientes. La incidencia de epilepsia es mayor en niños de entre 6 y 14 años, adolescentes y ancianos (la incidencia acumulada de epilepsia hasta la edad de 80 años alcanza el 3%) y, por sexos, mayor en varones. Se diagnostican entre 12.000 y 22.000 casos nuevos en España cada año (Serrano-Castro et al., 2015).
A nivel global, se estima que el 5-10% de la población experimentará una crisis a lo largo de su vida y hasta un 20% de estos tendrán crisis recurrentes, o sea, desarrollará epilepsia; poco más de la mitad de las crisis son parciales (57%) y más del 60% de los síndromes epilépticos son síndromes focales, más frecuentes en la edad adulta. La epilepsia incrementa entre 5 y 10 veces el riesgo de muerte prematura con la relación a la población general.
Desde un punto de vista clínico, las manifestaciones del cuadro epiléptico se relacionan con la localización anatómica del foco epiléptico y su extensión, y pueden ir desde un leve tic o un breve paréntesis en la atención del paciente a una crisis convulsiva generalizada, de varios minutos de duración. La ILAE clasifica las crisis epilépticas en función de la zona cerebral en que se originan y la forma de presentación –sintomatología motora y nivel de conciencia– en tres grandes grupos: de inicio focal, de inicio generalizado o de inicio desconocido (Figura 1).
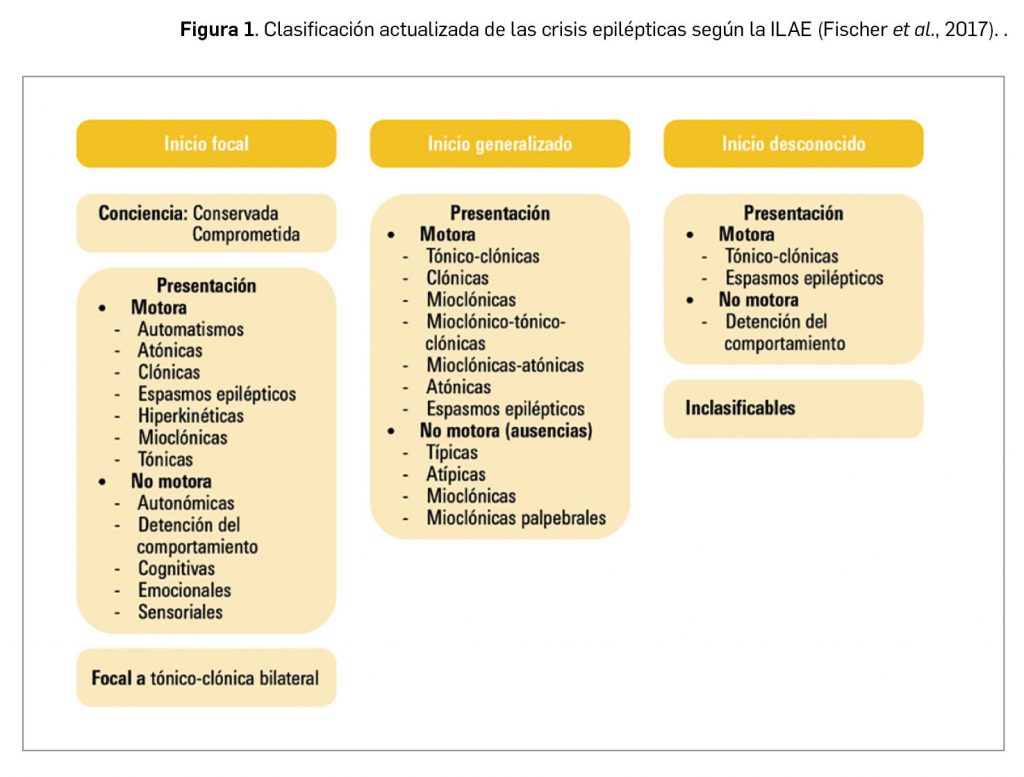
Las crisis epilépticas generalizadas suponen el 30% del total de casos diagnosticados. Se originan en un punto y rápidamente comprometen redes neuronales de distribución bilateral (en ambos hemisferios), que pueden incluir estructuras corticales y subcorticales, si bien no afectan necesariamente a toda la corteza cerebral, pudiendo ser asimétricas; pese a que una crisis individual pueda parecer localizada, la localización y lateralización no tienen por qué repetirse de una crisis a otra. Las crisis generalizadas pueden ser convulsivas, es decir, con movimientos asociados (tónicas, clónicas, tónico-clónicas y mioclónicas), o no convulsivas (ausencias y crisis atónicas).
Por otra parte, las crisis epilépticas focales –antes llamadas parciales– se originan en redes neuronales limitadas a un hemisferio, con una localización más o menos amplia. Para cada tipo de crisis, la localización de su inicio se repite de una crisis a otra, con patrones de propagación que pueden involucrar al hemisferio contralateral (secundariamente generalizadas). Suponen en torno al 60% de todos los cuadros de epilepsia y, según su localización, pueden ser frontales, parietales, temporales u occipitales; se categorizan como simples o complejas según haya o no afectación de la conciencia. Finalmente, los síndromes epilépticos agrupan a aquellos tipos de epilepsia que, teniendo formas de presentación y cursos clínicos similares, responden a una etiología diversa: cada tipo de síndrome epiléptico combina uno o más tipos de crisis epilépticas, con distintas manifestaciones.
Las causas de la epilepsia varían notablemente con la edad del paciente. En algunos tipos se presentan crisis en una etapa determinada de la vida y, con el tiempo, cesan; en otros, se producen crisis durante toda la vida. Un 30% de los casos de epilepsia son de origen genético, mientras que otro 40% están originados por malformaciones cerebrales, traumatismos craneoencefálicos, infecciones del sistema nervioso, hemorragias intracraneales, tumores cerebrales o trastornos metabólicos; el 30% restante son de causa desconocida.
Sea cual sea la causa inmediata, la base fisiopatológica de las crisis epilépticas es común: una descarga eléctrica neuronal anormal y exagerada producida por un desequilibrio a nivel sináptico de los impulsos nerviosos, los cuales están modulados fundamentalmente por la acción de dos neurotransmisores (uno de carácter neuroexcitatorio, el ácido glutámico, y otro neuroinhibitorio, el ácido gamma-aminobutírico o GABA). Según se ha sugerido, la epilepsia comporta la presencia de uno o varios núcleos de neuronas epileptógenas2 con una actividad eléctrica anormal y persistente en una zona cerebral que presenta una alteración del tejido nervioso y se denomina foco epiléptico. En otras palabras, las crisis epilépticas se producen por una activación sincrónica de uno o varios pequeños grupos de neuronas epileptógenas en el foco (se desencadena una actividad sináptica excitatoria excesiva) y puede difundir a otras áreas o incluso generalizarse.
Se ha descrito que muchos pacientes epilépticos desarrollan diversos mecanismos homeostáticos para contrarrestar la hiperexcitabilidad neuronal, probablemente relacionados con la expresión de varios genes que codifican distintos péptidos implicados en un aumento de la neurotransmisión GABAérgica o una disminución de la glutamatérgica. No obstante, en la gran mayoría de los casos se debe recurrir al tratamiento farmacológico. Aproximadamente dos tercios (50-70%) de los pacientes con epilepsia tienen buen pronóstico: además de un 20-30% de casos que remiten espontáneamente, otro 30-40% de los pacientes se controla con fármacos antiepilépticos; pero hasta un 20-30% muestran una persistencia de crisis en el tiempo. Se conoce la historia natural de algunos síndromes epilépticos (por ejemplo, la esclerosis mesial temporal), pero hoy en día el pronóstico de la epilepsia en el momento de su diagnóstico es indeterminado para muchos pacientes, lo cual se relaciona directamente con la probabilidad de conseguir estar sin crisis.
La farmacoterapia de las epilepsias a base de agentes antiepilépticos (o anticrisis) es crónico y se basa en controlar las crisis y mejorar las otras manifestaciones neurológicas asociadas a la patología, si bien no está exenta de efectos adversos que pueden ser fuente de discapacidad y morbilidad, lo que provoca bajas tasas de adherencia terapéutica a largo plazo (Fernández-Moriano, 2021). Iniciada a principios del siglo XX con el descubrimiento del fenobarbital, tiene un crecimiento notable a finales de ese siglo y principios del XXI, disponiéndose en la actualidad de un amplio abanico de opciones farmacológicas, con distintos perfiles farmacocinéticos y farmacodinámicos. Atendiendo a la hipótesis de la naturaleza electroquímica de la epilepsia, se pueden clasificar los fármacos antiepilépticos disponibles en dos grandes grupos según su actuación primordial sobre las neuronas del SNC:
- Inhibición de la hiperexcitabilidad neuronal cortical:
- Modulación de determinados canales iónicos de Na+, K+ y Ca2+ presentes en las membranas, responsables del intercambio iónico entre el interior y el exterior neuronal. Actúan sobre las propiedades eléctricas de las neuronas y su capacidad para ser excitadas por las descargas de otras neuronas.
- Inhibición del sistema glutamato-aspartato, implicado en la reducción del umbral de excitabilidad de las neuronas corticales.
- Inhibición de la exocitosis de neurotransmisores excitatorios (glutamato y aspartato) mediante la actuación sobre las glicoproteínas de la vesícula sináptica 2 (SV2)3, las cuales, presentes en las membranas neuronales, modulan la neurotransmisión normal regulando la exocitosis estimulada por Ca2+.
- Inhibición de la hiperexcitabilidad neuronal cortical:
- Potenciación del sistema GABAérgico. Los diferentes agentes que emulan la estructura a priori simple del GABA han mostrado gran complejidad farmacodinámica: actúan sobre la cinética del GABA o sus receptores por mecanismos diversos que frecuentemente coexisten para un mismo fármaco; existen agonistas de receptores GABAA y GABAB, potenciadores de la síntesis de GABA, inhibidores de su metabolismo o de su recaptación presináptica.
- Inhibición de la anhidrasa carbónica: esta acción, propia de algunos diuréticos (como acetazolamida), tiene interés en la epilepsia (sobre todo en cuadros refractarios) porque produce la acumulación de CO2 a nivel cerebral de la enzima localizada en la neuroglia, mielina y plexo coroideo; es la acidosis metabólica la que previsiblemente facilita la estabilización neuronal.
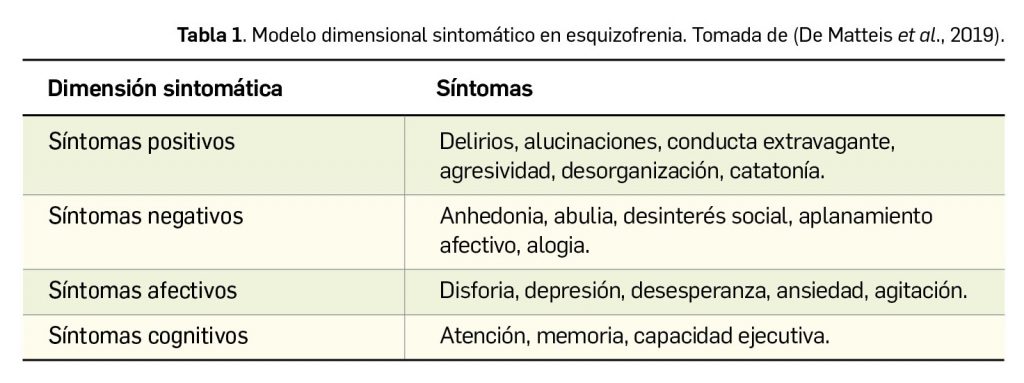
Como se ha sugerido, la gran mayoría de antiepilépticos actualmente usados en práctica clínica actúan a través de varios mecanismos al mismo tiempo con diferente relevancia en cuanto a sus efectos (Tabla 1). Esto, junto a las particularidades farmacocinéticas de cada uno de ellos, se traduce en una alta especificidad del perfil clínico de cada fármaco y determina que la elección de un tratamiento en concreto deba ser individualizada en función del perfil del paciente. Así, en ocasiones, las alternativas que puedan ser consideradas en un principio como de segunda elección por las guías clínicas pasan a ser la primera opción.
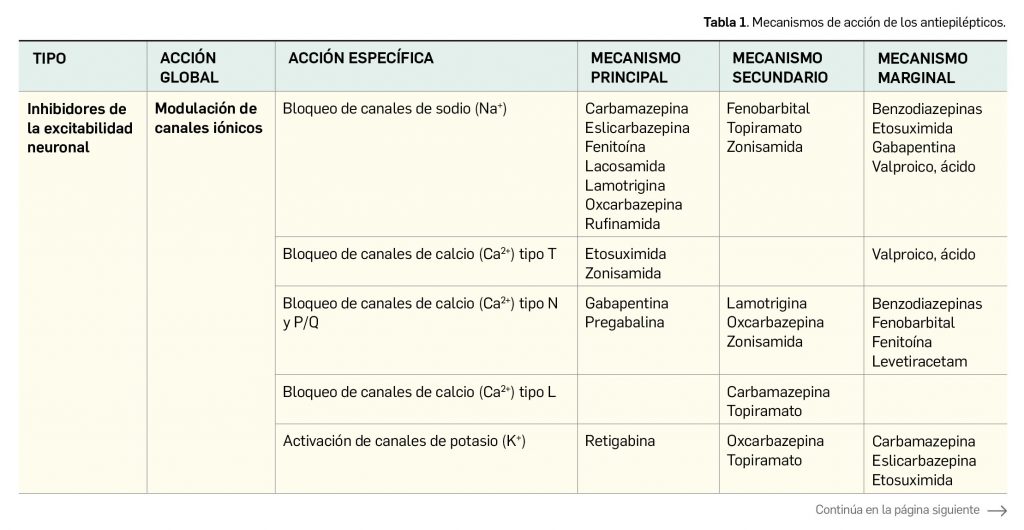
Continúa la tabla aquí
Es preciso subrayar que alrededor del 40% de los enfermos con una primera crisis tiene recidivas, por lo que el tratamiento no está indicado en todos los pacientes con una primera crisis; no se recomienda, por ejemplo, en las crisis agudas sintomáticas tras un traumatismo craneal o en niños con una primera crisis, pero sí en aquellas personas con mayor riesgo de recidiva, como pacientes con una patología cerebral adicional o antecedentes de epilepsia. En cambio, el riesgo de recidiva tras la segunda crisis asciende hasta casi el 70%: en esos casos el tratamiento está indicado casi siempre.
Hay consenso científico en torno a la idea de que el tratamiento debe iniciarse con un solo fármaco antiepiléptico, procediendo a titular la dosis hasta alcanzar la dosis eficaz o la dosis máxima tolerada. Si persisten las crisis, dicho fármaco debe ser sustituido por otro, añadiendo primero el segundo hasta alcanzar la dosis eficaz y, a continuación, reduciendo lentamente la dosis del primer fármaco hasta suspenderlo. No debe demorarse el cambio del fármaco una vez que se ha comprobado que las crisis persisten a pesar de emplear la dosis máxima tolerada. Numerosos ensayos clínicos controlados han registrado que en torno a un 60-65% de los pacientes responden bien al primer fármaco ensayado y un 20-25% adicional terminan respondiendo a la monoterapia mediante cambio de fármaco. Solo un 10% de los pacientes obtiene mayor beneficio de una combinación inicial de medicamentos que de un régimen en monoterapia. Por tanto, la idea prevalente en la actualidad es el cambio de medicación hasta encontrar el más adecuado y solo después de que se haya comprobado que la monoterapia es insatisfactoria, recurrir a las combinaciones.
El tiempo que debe mantenerse la farmacoterapia antiepiléptica es una cuestión delicada. Algunos pacientes necesitarán seguir la terapia de por vida, pero un porcentaje alto no recae cuando se suspende la medicación. Son factores desfavorables para una retirada del tratamiento –por aumentar el riesgo de recaída– presentar anormalidades neurológicas, ataques de más de un tipo, necesitar más de un medicamento o haber aparecido la epilepsia en edad adulta; pero cada caso requiere hacer la prueba. En niños que han pasado 2 años sin ataques, el índice de remisión es algo superior al 75%, mientras que en adultos es del orden del 60%. Muchos autores recomiendan una retirada muy gradual de la medicación una vez transcurridos 2 años sin ataques; otros elevan el tiempo de espera a 4 años.
En cualquier caso, la clave del tratamiento antiepiléptico reside en buscar un equilibrio entre efectividad y efectos adversos, ajustando la dosis para cada paciente y fármaco, a lo que ayuda en muchos casos la determinación de niveles plasmáticos, pues algunos fracasos de la terapia se atribuyen al abandono prematuro del fármaco por no obtener respuesta a corto plazo.
Habida cuenta de que la epilepsia es la 2ª patología neurológica en años de vida potencialmente perdidos o vividos con discapacidad, y de que la expectativa de vida de los pacientes se reduce entre 2 y 10 años, con una tasa de mortalidad entre 2 y 3 veces mayor que la población general4, resulta prioritario tratar aquellos casos de epilepsia farmacorresistente. La ILAE define la epilepsia farmacorresistente como “la persistencia de crisis cuando se han utilizado de forma adecuada, y a una dosis apropiada, dos fármacos anticrisis bien en monoterapia de forma secuencial o en combinación”. Pese a que se desconocen los motivos por los que en algunos pacientes no se controla la patología con medicamentos, sí se han descrito ciertos factores de riesgo, tales como: un mayor número de crisis antes de iniciar el tratamiento, respuesta inadecuada al tratamiento inicial con fármacos anticrisis o epilepsia focal estructural. Se sabe que en pacientes farmacorresistentes la epilepsia puede ser progresiva y tienen un mayor riesgo de daño cerebral y morbi-mortalidad, así como de consecuencias psicosociales negativas (exclusión social y laboral, accidentes, peor calidad de vida, etc.).
La mayoría de los ensayos clínicos que han sustentado la aprobación de los fármacos anticrisis de más reciente introducción se han realizado precisamente en personas con epilepsia focal farmacorresistente en tratamiento y a quienes se les añade el nuevo fármaco en estudio o placebo como tratamiento coadyuvante; a partir de ahí suelen derivarse investigaciones del fármaco en monoterapia u otros tipos de epilepsia. De esta forma se han autorizado perampanel (2012), acetato de eslicarbazepina (2012) y brivaracetam (2017).
En la actualidad, en base a los niveles de evidencia científica hay distintas pautas posibles de tratamiento tanto para la epilepsia focal como para las crisis generalizadas. La Sociedad Española de Neurología recomienda (grado de recomendación A) para el tratamiento de las crisis focales con o sin generalización secundaria en adultos el uso en monoterapia de carbamazepina, acetato de eslicarbazepina, gabapentina, lacosamida, levetiracetam, lamotrigina, oxcarbazepina, fenobarbital, fenitoína, topiramato, valproico o zonisamida. Como tratamiento adyuvante en regímenes dobles también recomienda el uso de brivaracetam, carbamazepina, clobazam, acetato de eslicarbazepina, gabapentina, lacosamida, levetiracetam, lamotrigina, oxcarbazepina, perampanel, pregabalina, topiramato y zonisamida. Algunos estudios han revelado que, a pesar de la disponibilidad de nuevos fármacos, hasta un 30-40% de los pacientes tendrán epilepsia refractaria a fármacos a pesar de una adecuada selección del tratamiento, lo que implicaría una cifra superior a 100.000 casos en España. Por eso, se necesitan nuevos fármacos que mejoren las tasas de respuesta de los fármacos que tenemos actualmente.
Acción y mecanismo
Cenobamato es un nuevo agente antiepiléptico con un mecanismo de acción dual: por un lado, actúa como modulador alostérico positivo del receptor del canal iónico del GABA (GABAA) mediante su unión a un lugar diferente al de las benzodiazepinas, y por otro, bloquea los canales de Na+, de forma que potencia la inactivación de estos canales e inhibe el componente persistente de la corriente de Na+, reduciendo la descarga neuronal repetitiva. Aunque no se conoce el mecanismo exacto que lo media, el fármaco se ha revelado eficaz en el control de las crisis epilépticas de inicio focal, de modo que el medicamento ha sido autorizado para el tratamiento concomitante por vía oral de las crisis de inicio focal con o sin generalización secundaria en adultos con epilepsia que no han sido controlados de forma adecuada a pesar del tratamiento previo con al menos 2 antiepilépticos.
Los estudios realizados durante su desarrollo preclínico y clínico han demostrado que la inhibición que cenobamato ejerce sobre la excitabilidad neuronal, es decir, su capacidad para hiperpolarizar el potencial de membrana es dosis-dependiente y más potente que el producido por lamotrigina y por carbamacepina (Nakamura et al., 2019).
Se ha probado que, sin mostrar una actividad agonista significativa, actúa como modulador al alza de la actividad de las corrientes iónicas inducidas por GABA en los 6 subtipos de receptores GABAA (α1β2γ2, α2β3γ2, α3β3γ2, α4β3γ2, α5β3γ2, α6β3γ2), con valores de EC50 que oscilan entre 42 y 194 μM (equivalente a 11-52 μg/ml). Su efecto no se alteraba por el antagonista del sitio de unión de las benzodiazepinas (flumazenilo), confirmándose así que la interacción del fármaco se produce en un sitio de unión distinto en el receptor GABAA. Además, los datos preclínicos sugieren no se produce tolerancia a los efectos anticonvulsivantes de cenobamato; por ejemplo, en las neuronas de ratas pretratadas durante 7 días no se afectaba la potenciación de GABAA por un nuevo tratamiento con el fármaco. Por otro lado, se ha visto en estudios in vitro con neuronas de rata que cenobamato reduce la amplitud de los picos de corrientes de sodio, tanto aquellas sensibles como resistentes a tetrodotoxina; esto sugiere que el fármaco puede actuar sobre una amplia variedad de canales de Na+, lo que se confirmó en estudios con isoformas humanas de canales de Na+, donde se probó un bloqueo eficaz en el estado inactivo de dichos canales con valores de CI50 entre 23 y 146 μM.
Conviene tener presente en los ajustes posológicos del tratamiento antiepiléptico que, por su capacidad de afectar al citocromo P-450 hepático, cenobamato puede interaccionar con otros antiepilépticos: puede reducir los niveles plasmáticos de fármacos metabolizados por CYP3A4 y CYP2B6 y aumentar la exposición a los fármacos metabolizados por CYP2C19. Esto se traduce, por ejemplo, en una potencial necesidad de reducir la dosis de fenitoína, fenobarbital o clobazam, y de aumentar la dosis de lamotrigina cuando se utilizan concomitantemente con cenobamato. No es necesario, en cambio, ningún ajuste posológico si se coadministra con carbamazepina, ácido valproico, lacosamida, levetiracetam u oxcarbazepina.
Aspectos moleculares
El nuevo fármaco tiene como nombre químico el de [(1R)-1-(2-clorofenil)-2-(tetrazol-2-il)etil] carbamato, que se corresponde con la fórmula molecular C10H10ClN5O2 y con la masa molecular relativa de 267,67 g/mol. Presenta una estructura química en la que destaca el anillo de tetrazol, que no se enmarcaría en ninguno de los grandes grupos de estructuras de los agentes antiepilépticos, como los ureidos y análogos, las carboxamidas o los análogos de GABA (véase Fernández-Moriano et al., 2021), pero que puede recordar parcialmente a algunos fármacos con predominio de heterociclos nitrogenados y átomos de halógenos, como rufinamida o lamotrigina (Figura 2).
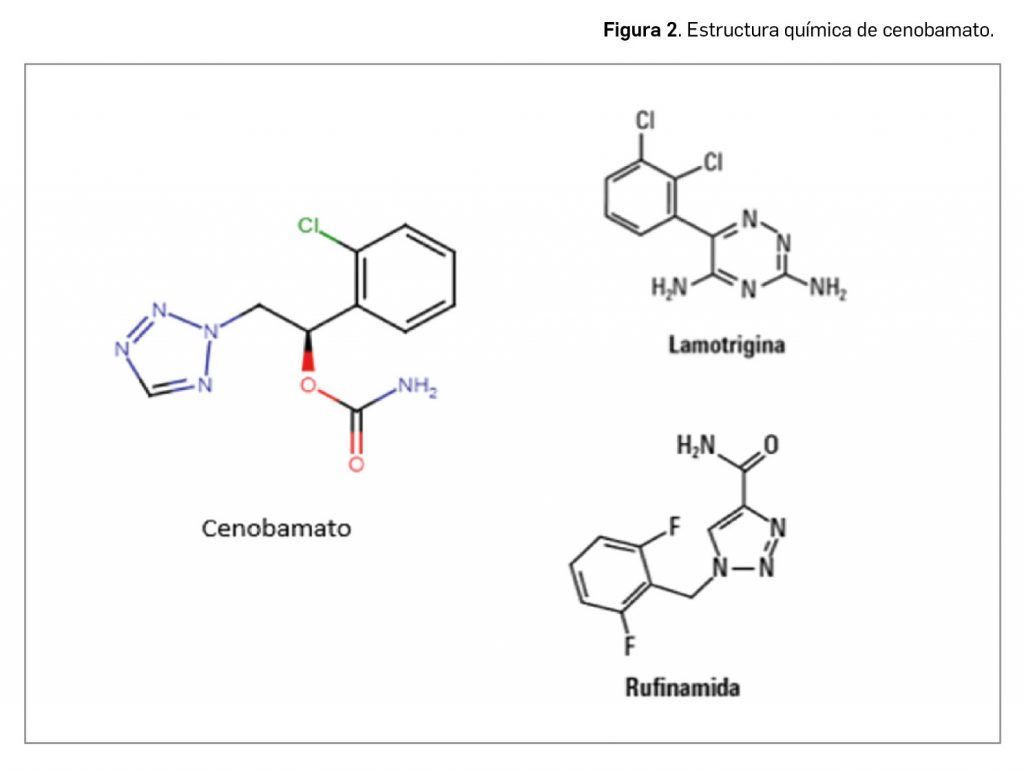
El principio activo se presenta como un polvo cristalino de color blanco o blanquecino, no higroscópico y ligeramente soluble en agua. La molécula exhibe estereoisomería debida a la presencia de un centro quiral, sintetizándose como el enantiómero R. También se ha observado polimorfismo, pero se sintetiza una única forma polimórfica de cenobamato.
Eficacia y seguridad clínicas
El balance beneficio-riesgo de cenobamato en su indicación y dosis autorizadas (12,5 mg/día al inicio, con aumento gradual hasta 200 mg/día) se contrastaron fundamentalmente en un ensayo pivotal de fase 2, multicéntrico y multinacional (107 centros de 16 países), con diseño aleatorizado, doble ciego y controlado por placebo, desarrollado con el objetivo principal de investigar la dosis óptima.
Dicho estudio C017 incluyó un total de 437 pacientes adultos (18-70 años) con epilepsia de inicio focal que no estaba bien controlada pese al uso previo –al menos en los últimos 2 años– de entre 1 y 3 antiepilépticos distintos, quienes fueron asignados al azar en 4 brazos paralelos a recibir bien cenobamato a una dosis de 100, 200 o 400 mg/día5 o bien un placebo equivalente. Se excluyeron pacientes con crisis psicógenas no epilépticas, epilepsia generalizada, síndrome de Lennox-Gastaut, antecedente de estatus epiléptico en los 3 meses previos o con una cirugía programada para su epilepsia; de igual modo, se excluyeron pacientes con otros trastornos psiquiátricos o en tratamientos con potencial de interacciones farmacológicas (por ejemplo, felbamato, diazepam, fenitoína, fenobarbital, vigabatrina o uso frecuente de benzodiazepinas de rescate).
Tras un periodo basal de 8 semanas en que los participantes debían presentar al menos 8 crisis con alteración motora o de la conciencia, o bien secundariamente generalizadas, fueron tratados con cenobamato durante 18 semanas, las 12 últimas a dosis fija. Es preciso subrayar que durante ese periodo se mantuvo estable el tratamiento con los otros antiepilépticos que cada paciente usara. Hasta el 74% de los pacientes tomaba ≥ 2 antitiepilépticos concomitantes al inicio, siendo los fármacos más comunes levetiracetam, lamotrigina, carbamazepina y lacosamida. Las características basales de los pacientes estuvieron equilibradas entre los grupos de tratamiento, destacando una mediana de edad inferior a 40 años y una mediana de crisis en el periodo basal de entre 8 y 11.
Los resultados de eficacia relativos a la población por intención de tratar modificada –ITTm– (n= 397; pacientes que habían completado la fase de titulación y tenían algún dato en el mantenimiento) tras 12 semana de mantenimiento a dosis fija se reflejan en la Tabla 2. Se consideró como variable primaria de eficacia la tasa de pacientes respondedores, entendiendo la respuesta como la reducción de ≥ 50% en el número de crisis durante la fase de mantenimiento del doble ciego respecto al periodo basal. Como variables secundarias se midieron el cambio porcentual en la frecuencia de crisis del periodo basal (tasa media mensual en 28 días) o las tasas de respondedores con criterios más estrictos (reducciones de ≥ 75%, ≥ 90% y del 100% en la frecuencia de crisis), entre otras.
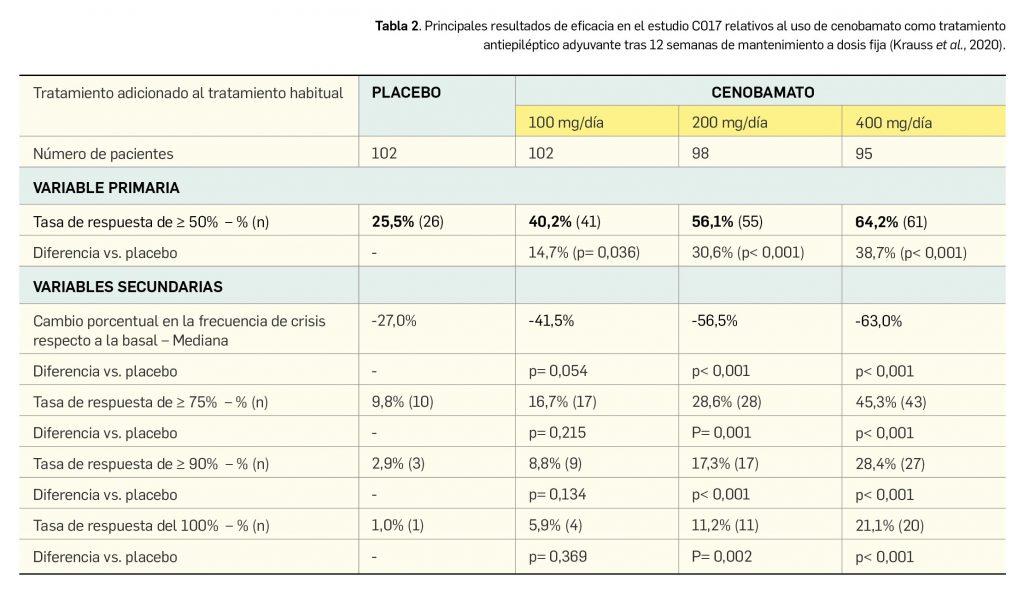
El análisis post-hoc por subtipos de crisis reveló que las tres dosis del fármaco redujeron significativamente durante la fase doble ciego el número de crisis focales sin alteración del nivel de conciencia. De modo interesante, las dos dosis más altas también redujeron notablemente la incidencia de crisis focales con alteración del nivel de conciencia y de las secundariamente generalizadas, que son las más graves y las que más comprometen la seguridad de los pacientes con epilepsia. Se observaron respuestas similares en los subgrupos de pacientes según la frecuencia de crisis basal (superior o inferior a la mediana) y según la duración de la enfermedad.
Además, la gran mayoría de pacientes (99%) eligió entrar en la fase de extensión abierta del estudio, de los cuales el 80% permaneció al menos 12 meses y el 58% durante 60 meses. En los primeros 6 meses de tratamiento, los pacientes que inicialmente habían sido asignados a placebo tuvieron una reducción media de crisis del 63%. A largo plazo se obtuvieron datos coherentes con los observados durante el periodo ciego, revelando que el efecto del nuevo fármaco perdura en el tiempo.
Por otra parte, un segundo ensayo de soporte (estudio C013), también de fase 2 y similar en diseño y desarrollo al pivotal, incluyó a un total de 222 pacientes con epilepsia farmacorresistente, historia de la enfermedad de al menos 2 años de duración y al menos 3 crisis focales mensuales pese al tratamiento estable (12 semanas previas) con 1, 2 o 3 fármacos anticrisis. Los pacientes fueron aleatorizados al azar a recibir cenobamato 200 mg/día (n= 113; dosis inicial de 50 mg/día y ascensos graduales) o un placebo equivalente (n= 106) durante un total de 12 semanas del doble ciego (6 de titulación y 6 de mantenimiento).
Al inicio del periodo basal la media de edad de los pacientes era de 39,6 años y tenían una mediana de crisis en 28 días de entre 5,5 (grupo de placebo) y 7,5 (grupo de cenobamato); en torno a la mitad de ellos (47-48%) estaba en tratamiento con 2 fármacos anticrisis y solo menos del 20% era tratado con monoterapia. La variable primaria de eficacia fue el cambio en el porcentaje de frecuencia de crisis en 28 días frente al periodo basal, mientras que el objetivo secundario era la tasa de respondedores (reducción ≥ del 50% en la frecuencia de crisis), medidos ambos a la semana 12.
Los resultados (Chung et al., 2020) revelaron en el grupo de cenobamato una disminución media de la frecuencia de crisis del 55,6%, desde 7,5 crisis en el periodo basal a 3,8 crisis en la fase doble ciego; esa reducción fue significativamente mayor (p< 0,0001) respecto al grupo placebo, donde el descenso medio fue del 21,5%, desde 5,5 a 5,0 crisis. La tasa de respondedores en el doble ciego fue también mayor en el grupo de cenobamato respecto al control, de 50,4% y 22,2% (p< 0,0001), respectivamente. Al final del tratamiento hasta un 8,8% de los pacientes tratados con el nuevo fármaco se mantuvo libre de crisis (tasa de respuesta del 100%), frente a solo el 0,9% en el grupo placebo; pero si se considera solo la fase de mantenimiento a dosis fija, esa proporción aumenta hasta el 28,3% con cenobamato y el 8,8% con placebo. En términos de seguridad, se acepta que el perfil toxicológico del nuevo fármaco ha sido bien caracterizado en hasta 26 ensayos clínicos que han incluido a más de 2.500 sujetos.
Los datos más relevantes proceden de los dos estudios citados y de otro ensayo abierto de fase 3 (estudio C021; N= 1.340) desarrollado específicamente para evaluar la seguridad del fármaco en términos de incidencia de DRESS (siglas del inglés “reacción de sensibilidad cutánea a medicamentos con eosinofilia y síntomas sistémicos”), ante el riesgo que se advirtió con el uso del fármaco a dosis altas de inicio y titulaciones rápidas (Sperling et al., 2020)6. En todos ellos se vio que los efectos adversos más frecuentes (> 10%) relacionados con cenobamato son aquellos sobre el sistema nervioso central, tales como son somnolencia, mareo, fatiga y cefalea; otros menos frecuentes (< 10%) fueron cuadros confusionales, irritabilidad, diplopía, nistagmo, afasia y alteración de la memoria, e incluso menos frecuentes fueron la ataxia y los trastornos gastrointestinales. En los estudios controlados se vio una incidencia de eventos adversos mayor con cenobamato que con placebo (77% vs. 68%), con tasas de interrupción por motivos de seguridad de entre el 6-19% (vs. 3%); la dosis de 400 mg/día se asoció con una peor tolerabilidad, en especial cuando se tomaba de forma simultánea con clobazam.
Sin embargo, en su mayoría son eventos adversos leves-moderados en severidad y autolimitados en duración, tienen una incidencia dependiente de la dosis y de la velocidad de titulación (se vieron casos más graves cuanto más rápido se incrementó la pauta). La tasa de eventos adversos graves ronda el 14%, entre los cuales la ataxia y el mareo fueron los que provocaron más casos de discontinuación del tratamiento (1,6% en cada caso vs. 0,5% con placebo), seguidos de somnolencia (1,4% vs. 0,5%). Se acepta que esas reacciones adversas son más incidentes en la fase de titulación, ya que en la fase de mantenimiento se asemejan al grupo placebo, incluso a la dosis de 400 mg/día.
Aspectos innovadores
Cenobamato es un nuevo agente antiepiléptico cuyo mecanismo de acción no ha sido del todo dilucidado, si bien se ha postulado que ejerce una acción dual: actúa como modulador alostérico positivo del receptor del canal iónico del GABA (GABAA) mediante su unión a un lugar diferente al de las benzodiazepinas, y bloquea e inactiva de forma más prolongada los canales de Na+, inhibiendo el componente persistente de la corriente iónica. En consecuencia, consigue reducir la excitabilidad neuronal y la descarga repetitiva. Por su eficacia en el control de las crisis epilépticas, el medicamento ha sido autorizado para el tratamiento concomitante por vía oral de las crisis de inicio focal con o sin generalización secundaria en adultos con epilepsia que no han sido controlados de forma adecuada a pesar del tratamiento previo con al menos 2 antiepilépticos.
Los datos clínicos que sustentaron su aprobación proceden fundamentalmente de un estudio pivotal aleatorizado de fase 2 (N= 437), de búsqueda de dosis y controlado por placebo, en que cenobamato se adicionó a la terapia previamente establecida. Sus resultados probaron que, tras un periodo inicial de 6 semanas de titulación y otras 12 semanas de mantenimiento a dosis fija, la tasa de respuesta con cenobamato –reducción de la frecuencia de crisis ≥ 50%7– fue notablemente más alta que con placebo: 56% con la dosis de 200 mg/día y 64% con 400 mg/día (dosis máxima), frente a una tasa del 27% con placebo; hasta el 11% y el 21% de pacientes se mantuvieron libres de crisis con ambos niveles de dosis (vs. 1% con placebo). De modo interesante, se vio que las dos dosis citadas de cenobamato reducían sustancialmente la incidencia de crisis con alteración del nivel de conciencia y de crisis focales que progresan a bilaterales tónico-clónicas (las más graves). La eficacia del fármaco se mostró consistente en los distintos subgrupos de pacientes, con independencia del tratamiento previo y concomitante, y se mantenía en el tiempo: se observaron resultados similares tras al menos 25-30 meses de mantenimiento en la fase de extensión abierta (reducción de la frecuencia de crisis del 76%, con un 20% de los pacientes libres de crisis).
Un segundo estudio de soporte (N= 222) –también de fase 2 y similar diseño– obtuvo resultados coherentes con los del pivotal. Tras 12 semanas de adición de cenobamato –6 semanas a dosis fija de 200 mg/día– al tratamiento establecido (monoterapia o, más frecuentemente, la combinación de dos antiepilépticos) se apreció una reducción de la frecuencia de crisis epilépticas del 56% (desde 7,5 crisis en el periodo basal a 3,8 crisis en la fase doble ciego), significativamente mayor que con placebo (-22%, desde 5,5 a 5,0 crisis). La tasa de respondedores fue también superior con cenobamato (50% vs. 22%), con una proporción de pacientes libres de crisis de hasta el 28% (vs. 9%) si se considera solo la fase de mantenimiento.
En términos de seguridad, los datos de esos dos estudios, complementados con un fase 3 más amplio, ponen de manifiesto un perfil toxicológico dependiente de la pauta posológica, predecible (similar al de otros antiepilépticos) y relativamente benigno para el nuevo fármaco, con una incidencia de reacciones adversas solo ligeramente superior a placebo (77% vs. 68%). Por su frecuencia (> 10%) destacan las relacionadas con el sistema nervioso central, como somnolencia (mayor riesgo en coadministración con benzodiazepinas), mareo, fatiga y cefalea; la mayoría de ellas son leves-moderadas y de corta duración. La incidencia de eventos adversos graves ronda el 14% y se asocia con una tasa de interrupción variable que puede alcanzar el 19%, siendo la ataxia, los mareos y la somnolencia las principales causas (< 2% en todos los casos). No obstante, la alta tasa de retención del fármaco en los ensayos de extensión (> 70% a los 5 años) es un buen indicador tanto de su eficacia como de su seguridad a largo plazo. Además, parece confirmado que una titulación lenta del fármaco desde una dosis inicial baja permite mitigar el riesgo de DRESS.
Entre las principales limitaciones de la evidencia hasta ahora disponible sobresale el diseño de fase 2 de los estudios principales y, en especial, el uso de placebo como comparador, que limita la duración de los estudios por razones éticas. En cualquier caso, esta circunstancia se acepta por la necesaria evaluación de la seguridad del fármaco, motivo por el cual la autorización de los fármacos anticrisis más recientes se ha basado en estudios controlados por placebo; no existe a día de hoy un fármaco considerado comparador de referencia en la epilepsia focal del adulto (AEMPS, 2022). Tampoco se dispone de datos suficientes respecto al uso de cenobamato en adultos mayores de 65 años.
En ausencia de comparaciones directas con otros antiepilépticos, si se hiciera una comparación indirecta –de robustez estadística limitada– frente a otros ensayos con diseños y poblaciones de similares características, destacaría la tasa de pacientes libres de crisis con cenobamato, que puede superar el 20% y es más relevante que la observada en los estudios clínicos con otros fármacos anticrisis usados en epilepsia focal, para los que esa proporción no supera el 7%. Tal afirmación sería extensible a fármacos como lacosamida, eslicarbazepina, levetiracetam, tiagabina, zonisamida, pregabalina, gabapentina, topiramato, lamotrigina y oxcarbazepina (Costa et al., 2011), pero también para los antiepilépticos más recientes como brivaracetam o perampanel, que rondan el 5%. En las comparaciones indirectas también merecerían una reseña las altas tasas de retención con cenobamato, que en los primeros 2 años de tratamiento rondan el 70% y serían similares o superiores a otros antiepilépticos (Kwok et al., 2017), pero que, sobre todo, se mantienen estables a largo plazo (casi un 60% a los 6 años), cuando serían superiores a las descritas para otros fármacos anticrisis como lacosamida (37% a los 3 años), perampanel (43%) y levetiracetam (58%) (AEMPS, 2022).
A fin de contextualizar el posicionamiento de cenobamato, se debe recordar que, pese a los importantes avances en las 3 últimas décadas en las posibilidades de individualización de la farmacoterapia antiepiléptica, no existe para esta compleja patología un único tratamiento estándar de elección y, en especial, que ese progreso no ha tenido la misma repercusión en el 20-30% de los pacientes que tienen epilepsia refractaria. Se estima que en España hay en torno a 54.000 pacientes con epilepsia de inicio focal resistente a al menos 2 fármacos, quienes habrán sido tratados en una primera línea en monoterapia y en una segunda con otra opción de monoterapia o con una primera adyuvancia; muy probablemente, habrán usado un antiepiléptico más reciente (los llamados de 3ª generación), como brivaracetam, perampanel, eslicarbazepina o lacosamida, que suelen tener un mejor perfil de efectos adversos y de interacciones farmacológicas.
Según recoge la Sociedad Española de Neurología entre sus recomendaciones, es precisamente la población de adultos con crisis de inicio focal farmacorresistentes –que no hayan conseguido un control adecuado o no toleren las líneas previas– la que se podrá beneficiar del uso diario por vía oral del nuevo fármaco como alternativa de tratamiento adyuvante en una 3ª línea como primera o segunda adyuvancia. En definitiva, cenobamato no incorpora un mecanismo de acción novedoso en la indicación ni parece suponer un cambio de paradigma en el abordaje de la epilepsia, pero sí va a cubrir una laguna terapéutica por tratarse del primer medicamento específicamente autorizado para el tratamiento concomitante tras el fallo de 2 o más fármacos. Sin embargo, su lenta titulación (puede tardarse 2-3 meses en alcanzar la dosis deseada) y el hecho de no estar disponible en formulación intravenosa impiden su empleo en situaciones de urgencia. Serán los resultados de futuros estudios los que determinen si se puede usar el fármaco en monoterapia, en líneas anteriores de tratamiento o en otros tipos de epilepsia.
Valoración