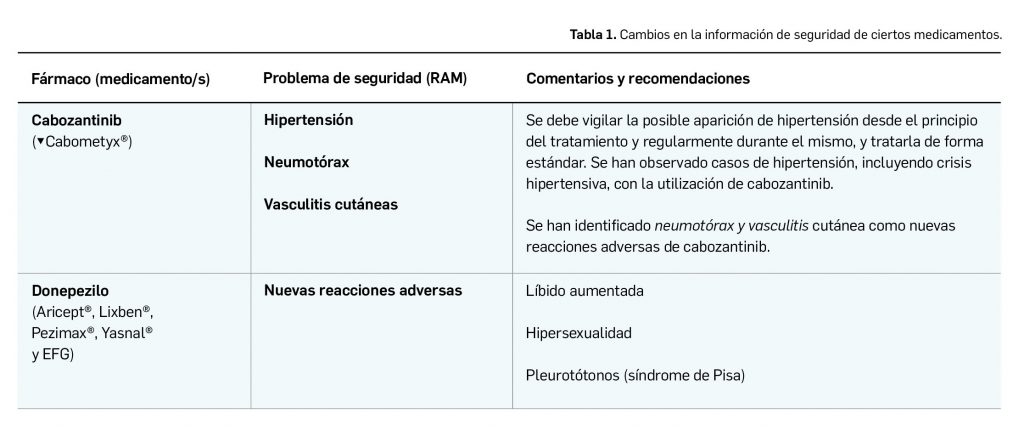
La agencia reguladora de medicamentos del Reino Unido, la MHRA, ha informado del riesgo de niveles bajos de vitamina B12 en el 10% de los pacientes diabéticos tratados con metformina.
En el Reino Unido, su agencia reguladora de medicamentos (Medicines and Healthcare products Regulatory Agency, MHRA) ha comunicado que la información de los medicamentos con metformina se ha actualizado para indicar que la deficiencia de vitamina B12 es una reacción adversa común a la metformina y puede afectar hasta 1 de cada 10 personas que la toman (MHRA, 2022).
Metformina está indicada –y es ampliamente usada– para el tratamiento de la diabetes mellitus tipo 2 y la prevención de la diabetes tipo 2 en pacientes con alto riesgo de desarrollarla. La deficiencia de vitamina B12 es una reacción adversa conocida a la metformina, y la literatura actual ha sugerido que la frecuencia de esta reacción adversa es más alta de lo que se pensaba anteriormente.
La información del medicamento también se actualizó para señalar que el riesgo de esta reacción adversa se incrementa con el aumento de la dosis de metformina y la duración del tratamiento, y en pacientes con factores de riesgo que se sabe que causan deficiencia de vitamina B12.
Recomendaciones
Se recomienda a los profesionales sanitarios que analicen los niveles de vitamina B12, en aquellos pacientes que presentan anemia o neuropatía, y que se debe considerar el control periódico de vitamina B12 en pacientes con factores de riesgo de deficiencia de vitamina B12.
En nuestro ámbito, se debe recordar que en la información contenida en las fichas técnicas de los medicamentos con metformina autorizados (Brotmin®, Dianben®, Ubene® y EFG), en la sección 4.4 Advertencias y precauciones especiales de empleo, se incluye la información siguiente (AEMPS, 2022):
La metformina puede reducir los niveles séricos de vitamina B12. El riesgo de disminución de la vitamina B12 aumenta con la dosis de metformina, la duración del tratamiento y/o en pacientes con factores de riesgo conocidos que causen deficiencia de vitamina B12. En caso de sospecha de deficiencia de vitamina B12 (como anemia o neuropatía), los niveles séricos de vitamina B12 deben ser monitorizados. Puede ser necesaria la monitorización periódica de los niveles séricos de vitamina B12 en pacientes con factores de riesgo para la deficiencia de vitamina B12. El tratamiento con metformina debe continuar en la medida en que este fármaco sea tolerado y no esté contraindicado y, además, se administre el tratamiento correctivo de la deficiencia de vitamina B12 apropiado de acuerdo con las guías clínicas vigentes.