Número 456, Septiembre 2022
Resumen de las notas sobre seguridad y farmacovigilancia publicadas por la AEMPS desde principios del año 2022. Para información más ampliada y acceso al documento de la AEMPS, puede consultar BOT PLUS.
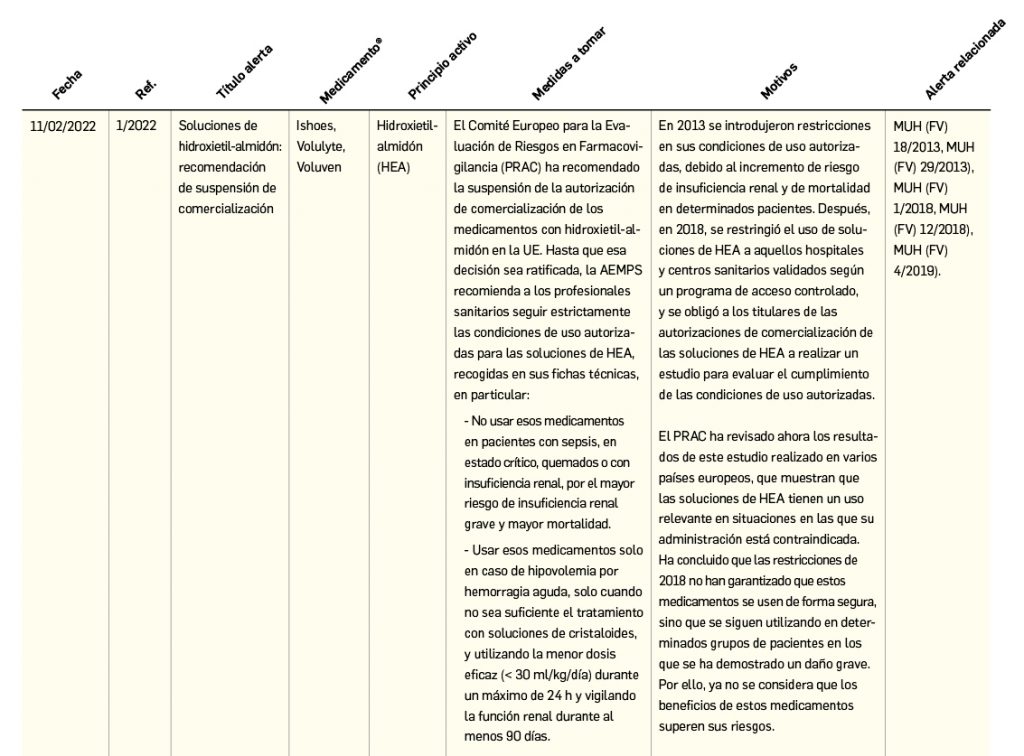
Alertas debidas a defectos de calidad observados en medicamentos de uso humano, publicadas por la AEMPS desde el anterior número y que suponen la retirada o inmovilización de ciertos lotes de medicamentos. En BOT PLUS puede encontrar más información detallada, con acceso al documento de la AEMPS.
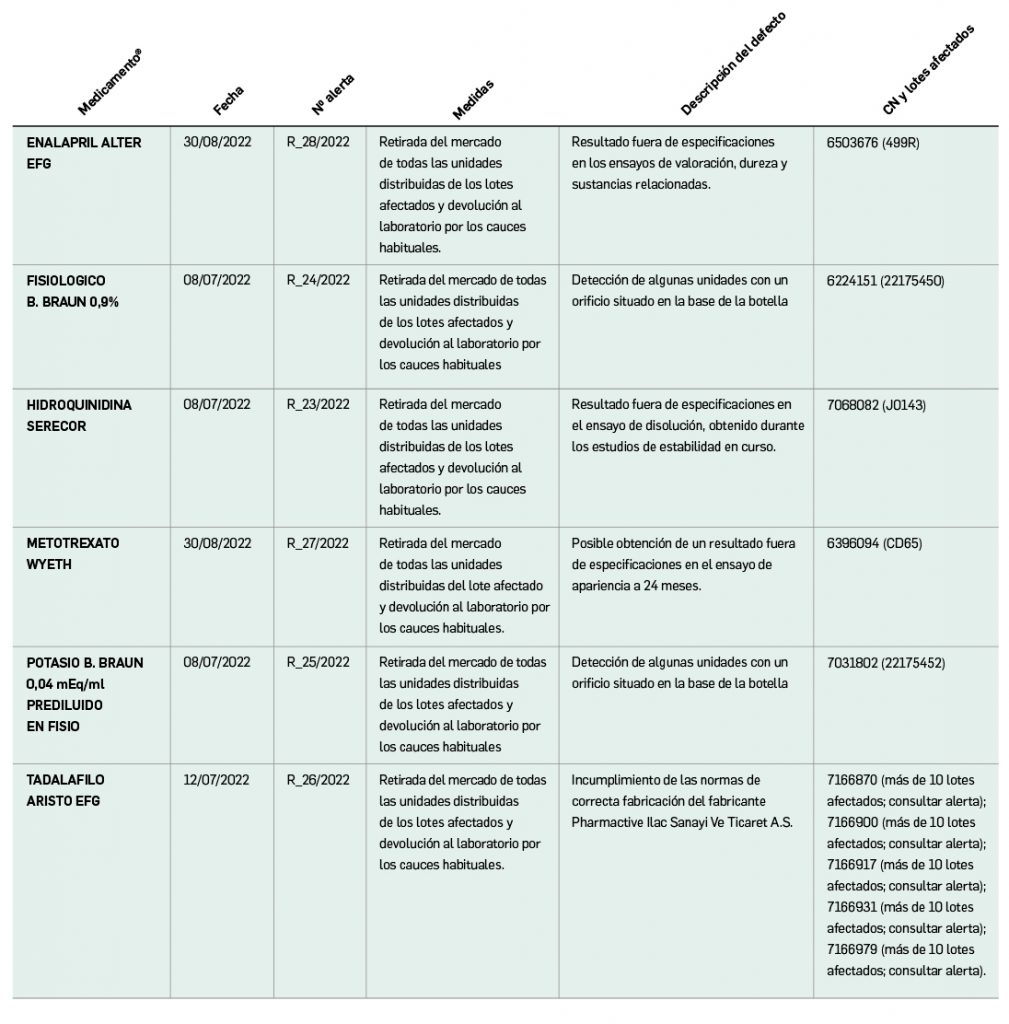
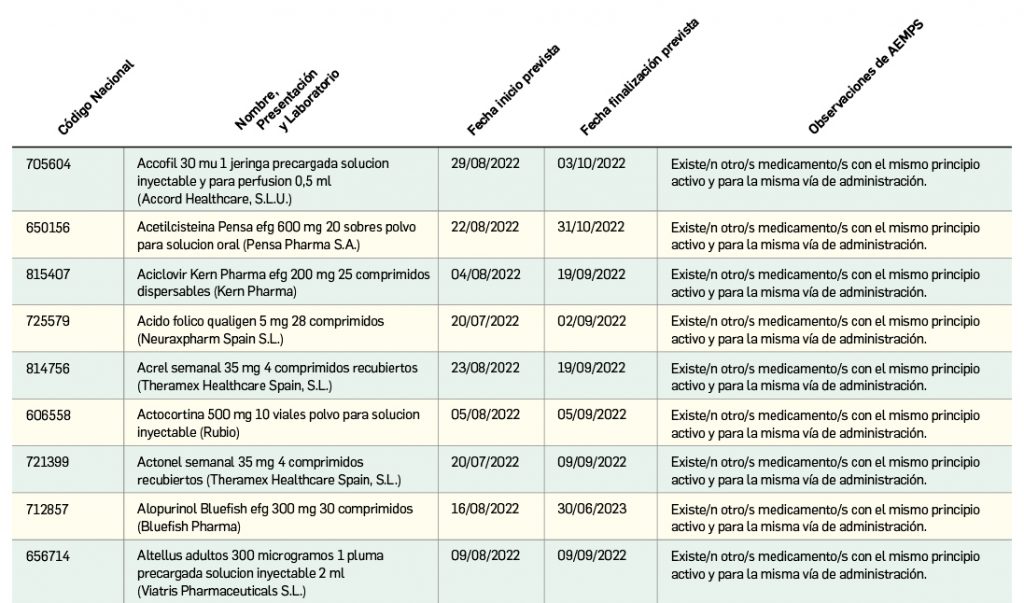
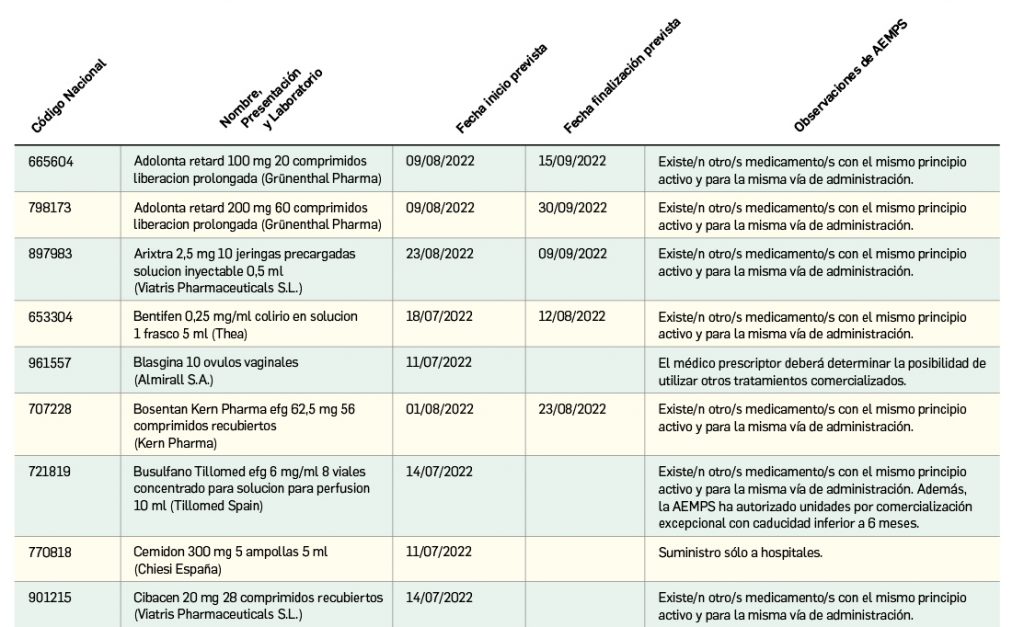
Listado de medicamentos con problemas de suministro publicado por la AEMPS, a fecha de cierre de este número. En BOT PLUS se puede encontrar la información completamente actualizada, al tratarse de una información que varía de forma continua.
Los medicamentos de terapia avanzada (MTA o Advanced Therapy Medicinal Products, ATMP) ofrecen nuevos e innovadores tratamientos para las enfermedades. Están basados en la terapia génica, la terapia celular somática o la ingeniería tisular. El marco legal para las ATMP en la Unión Europea está establecido en la Regulation (EC) No 1394/2007 on advanced therapy medicinal products que asegura el libre movimiento de estas medicinas dentro de la Unión Europea y el acceso a los mercados. La regulación (EC) nº 1394/2007 también establece el nuevo Comité en Terapias avanzadas (CAT) cuya responsabilidad fundamental consiste en preparar un proyecto de opinión sobre cada nueva solicitud de medicamento de terapia avanzada planteada a la Agencia Europea de Medicamentos, antes de que el Comité de Medicamentos de Uso Humano (CHMP, Committee for Medicinal Products for Human Use) de la misma adopte una opinión definitiva sobre la concesión, modificación, suspensión o revocación de una autorización de comercialización para el medicamento en cuestión.
La siguiente tabla recoge los medicamentos de terapia avanzada que han recibido autorización de comercialización en la UE durante en los últimos 10 años (2012-2022).
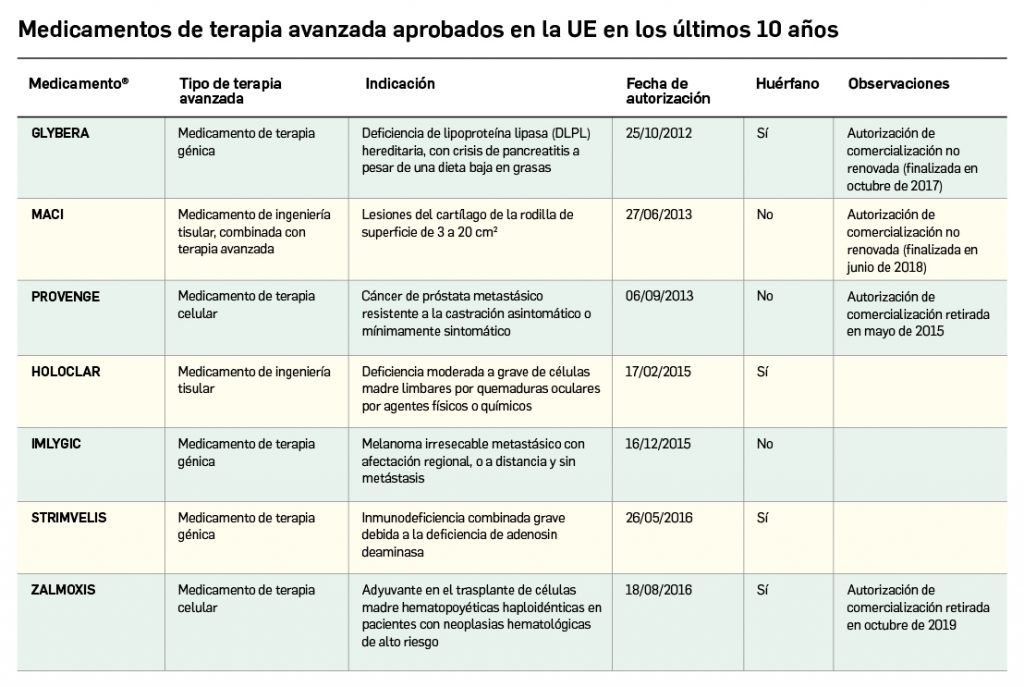
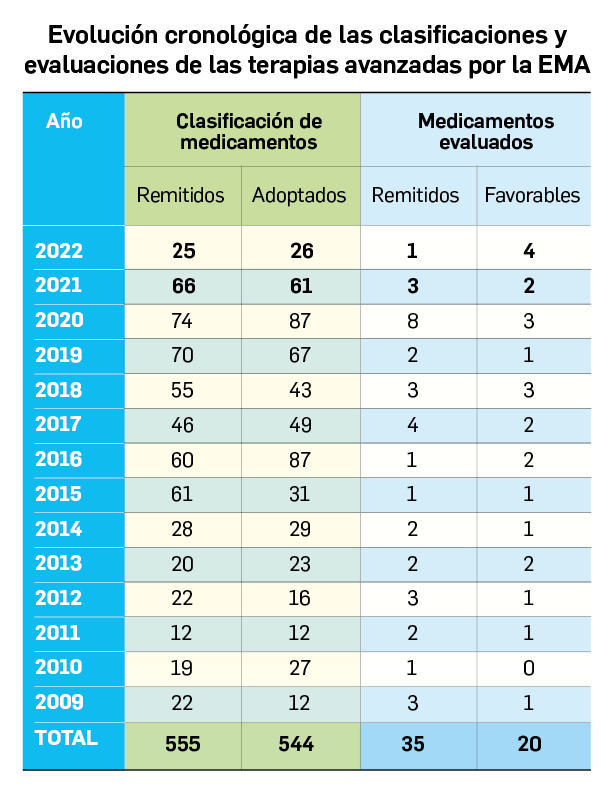
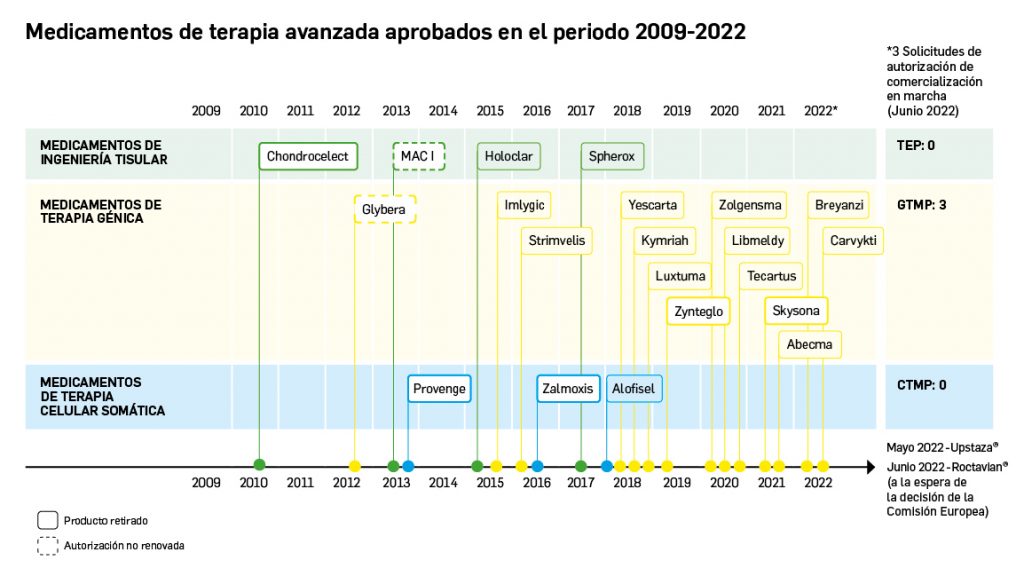
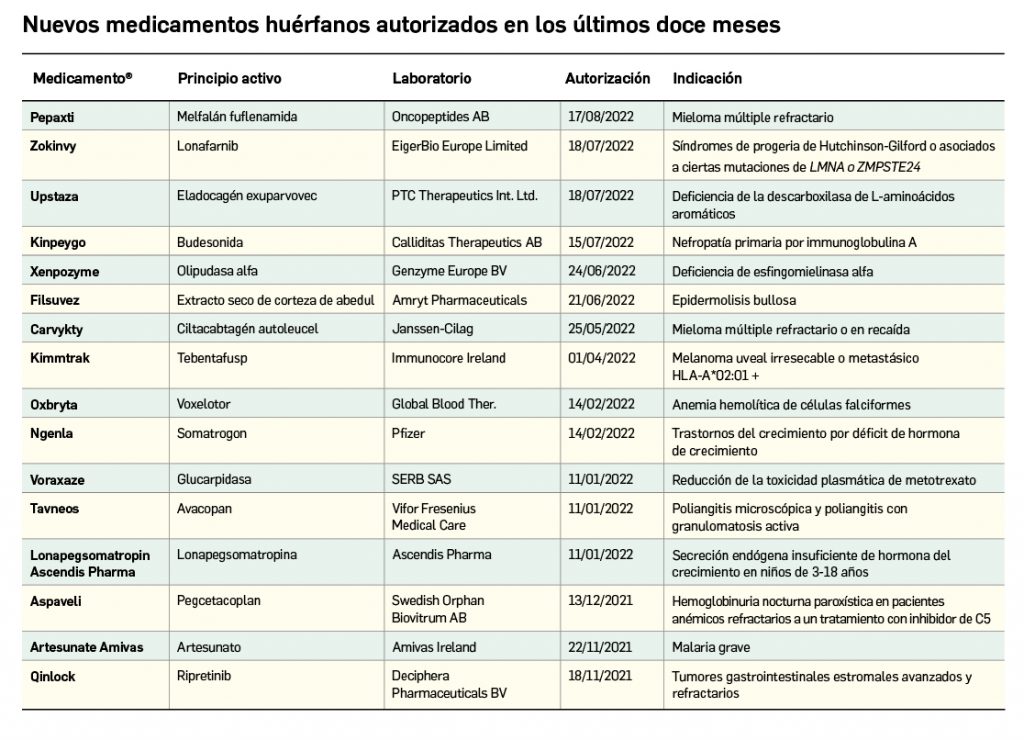
Los medicamentos huérfanos son aquéllos que sirven para diagnosticar, prevenir o tratar enfermedades raras de carácter muy grave o con riesgo para la vida. En la Unión Europea, la calificación de enfermedad rara se aplica a todas aquellas que no afectan a más de 5 de cada 10.000 personas. La designación de un medicamento como huérfano no garantiza su uso en la condición designada y no implica necesariamente que el producto satisfaga los criterios de eficacia, seguridad y calidad necesarios para la concesión de la autorización de comercialización. Como para cualquier medicamento, estos criterios solo pueden ser evaluados una vez que la solicitud de autorización de comercialización haya sido presentada.
► Agencia Europea de Medicamentos (EMA; Europea Medicines Agency). Apartado de Medicamentos Huérfanos
(inglés): https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview
https://www.ema.europa.eu/en/committees/committee-orphan-medicinal-products-comp
► Comisión Europea: web oficial de la Comisión Europea sobre enfermedades raras y medicamentos huérfanos (español).
http://ec.europa.eu/health/rare_diseases/policy/index_es.htm
► Orphanet: Portal de información oficial de la Unión Europea sobre enfermedades raras y medicamentos huérfanos (español).
http://www.orpha.net/consor/cgi-bin/index.php?lng=ES
► Eurordis: Federación Europea de Asociaciones de Pacientes con Enfermedades Raras (español). http://www.eurordis.org/es
► Food & Drug Administration (FDA, Estados Unidos). Apartado de Medicamentos Huérfanos (inglés):
http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/default.htm
► Pharmaceuticals & Medical Devices Agency. Agencia de Medicamentos y Dispositivos Médicos, de Japón (inglés):
http://www.pmda.go.jp/english/index.html
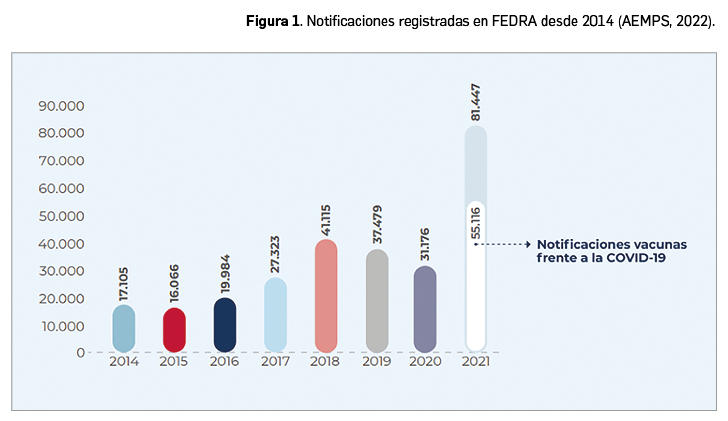
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha publicado el resumen de las actividades desarrolladas por el Sistema Español de Farmacovigilancia Humana (SEFV-H) durante el año 2021, el segundo año de la pandemia de la COVID-19. Durante este periodo se ha llevado a cabo en España la vacunación masiva frente al SARS-CoV-2, lo que se ha reflejado en la actividad del SEFV-H: de las 81.447 notificaciones de sospechas de reacciones adversas a medicamentos que se han recibido, el 67,7% correspondieron a acontecimientos adversos ocurridos tras la administración de alguna de las vacunas autorizadas frente a la COVID-19.
La AEMPS agradece la colaboración de los profesionales sanitarios y de la ciudadanía, y recuerda la importancia de notificar cualquier sospecha de reacción adversa (RAM) o, en el caso de las vacunas, de cualquier acontecimiento adverso tras la vacunación. Esta información es muy relevante para identificar potenciales nuevos riesgos de los medicamentos para su posterior análisis y toma de medidas.
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha comunicado el resumen de actividades del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano (SEFV-H) correspondiente al año 2021 (AEMPS, 2022).
En ese año se registraron en FEDRA (base de datos común para el SEFV-H) un total de 81.447 notificaciones de sospechas de reacciones adversas a medicamentos (RAM) o de acontecimientos adversos tras recibir una vacuna (ESAVI, eventos supuestamente atribuibles a la vacunación o inmunización), lo que ha supuesto un incremento de un 161% respecto al año anterior. Del total de notificaciones recibidas (81.447 casos), 55.116 se relacionan con acontecimientos adversos tras recibir alguna de las vacunas frente a la COVID-19. En concreto, dos tercios, el 67,7% de todos los casos notificados al SEFV-H han sido ESAVI, ya que son RAM pero que pueden ser explicadas por la vacuna o bien por otros factores coincidentes en el tiempo, y no tener relación causal, sino casual. En la Figura 1 se muestra el incremento que presentan los últimos años, y en conceto en 2021, en el número de notificaciones registradas en FEDRA desde 2014.
El 74,7% de casos (60.829 casos) fueron notificados directamente al SEFV-H, la mayor parte por profesionales sanitarios (46%), seguido del 25% procedentes de la ciudadanía directamente, 16% notificados por profesionales de enfermería, 9% de profesionales farmacéuticos, y un 3% por profesionales que no se especifican en la notificación. El 25,01% del total de casos notificados en 2021 procedían de los laboratorios farmacéuticos titulares de los medicamentos sospechosos de los casos de RAM que les comunicaron los profesionales sanitarios. El restante 0,2% fueron casos de sospechas de RAM (ICSR) recibidos a partir de la revisión de casos de RAM publicados en las revistas biomédicas, que lleva a cabo la Agencia Europea de Medicamentos (EMA), conocido como el servicio Medical Literature Monitoring, o MLM Service.
De los casos notificados al SEFV-H (60.829 casos) un 99% fueron notificaciones espontáneas –farmacovigilancia pasiva– enviadas a los centros autonómicos. Para los casos notificados a través de la industria farmacéutica, por profesionales o ciudadanos, este porcentaje fue un 59%. El resto de los casos procedían de sistemas de recogida de información organizada (farmacovigilancia activa), tales como estudios observacionales, programas de seguimiento de pacientes o acceso al medicamento a través del uso compasivo.
Los casos comunicados considerados “graves” correspondían al 23% de los recibidos en los centros del SEFV-H, y al 30% de los casos comunicados a través de la industria farmacéutica.
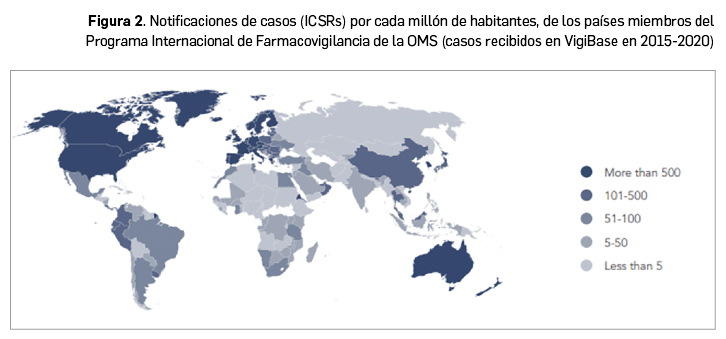
La tasa de notificación global en 2021 se estima en 128 casos por cada 100.000 habitantes, o 1.280 casos por millón de habitantes, tal como se expresan los datos globales del Programa Internacional de Farmacovigilancia de la OMS, conocido como PIDM, por sus siglas en inglés, coordinado desde el Uppsala Monitoring Centre (UMC). En el entorno mundial es una tasa de notificación alta, como se aprecia en la Figura 2. En los datos acumulados de los casos individuales de RAM, conocidos en inglés como ICSR (Individual Case Safety Reports), recibidos entre 2015 y 2020 en la base de datos VigiBase, se puede constatar que la tasa de 1.280 ICSR por millón corresponde al primer grupo de países que aportan anualmente más de 500 casos por millón (UMC, 2020).
No se debe olvidar que la calidad, y no tanto la cantidad, es la fuente principal de datos de seguridad de medicamentos, importante en farmacovigilancia.
En cuanto a la naturaleza de las sospechas de reacciones adversas notificadas durante el 2021, los trastornos generales, del sistema nervioso y del sistema músculo-esquelético fueron los más frecuentes.
Revisando los datos del SEFV-H se constata que un 29% de los 60.829 casos notificados, describían RAM desconocidas para el medicamento que se consideró sospechoso, tomando como referencia a su ficha técnica. Las notificaciones “graves”, que además eran “desconocidas”, fueron un 13%. Estos casos tienen un gran valor para la generación de “señales”, que se definen como potenciales nuevas reacciones adversas que deben seguir investigándose. Estas se han revisado exhaustivamente por los técnicos del SEFV-H con un análisis de forma continua y prioritaria para detectar si existe un riesgo potencial no conocido que necesite una evaluación más profunda, la cual se lleva a cabo en el seno del Comité Europeo para la Evaluación de Riesgos en Farmacovigilancia (PRAC, por sus siglas en inglés), mediante modelos matemáticos que indican si se están notificando más acontecimientos de los que cabría esperar en base a las incidencias basales de determinadas enfermedades calculadas en ausencia de vacunación.
El SEFV-H ha contribuido en la identificación de importantes asuntos de seguridad como han sido, entre otros, el “síndrome de trombosis con trombocitopenia” [Notas Informativas MUH (FV), 04/2021 y MUH (FV), 07/2021] o el síndrome de Guillain-Barré [Notas Informativas MUH (FV), 13/2021 y MUH (FV), 14/2021] para las vacunas de adenovirus (Vaxzevria® y COVID-19 Vaccine Janssen®).
La crisis sanitaria provocada por la COVID-19 sigue teniendo un alto impacto en las actividades del SEFV-H, que se ha ido adaptando en cada momento a las necesidades surgidas desde el inicio de la pandemia en el primer trimestre de 2020. Así, una vez autorizada la primera vacuna e iniciada la campaña de vacunación a finales de 2020, el trabajo del SEFV-H se ha enfocado durante el año 2021 en el registro y análisis de los acontecimientos adversos notificados. Ya antes del inicio de la vacunación la AEMPS estableció un plan de vigilancia para estas vacunas, intensificando algunas de las actividades en farmacovigilancia y el primer paso ha sido analizar los acontecimientos adversos que se registran, siendo esta la actividad prioritaria del SEFV-H a lo largo de este año, llevándose a cabo una serie de actividades para resaltar la importancia de la notificación, facilitar dicha tarea y priorizar el análisis, como:
En el año 2020 se observó una disminución del 17% en el número de notificaciones recibidas respecto al año 2019. En el año 2021 debido a la alta presión asistencial originada por la pandemia, se ha aumentado la notificación en un 161%. Desde el inicio de la campaña de vacunación hasta el 31 de diciembre del 2021 se han administrado alrededor de 73 millones de dosis de vacunas, lo que correspondería aproximadamente a 75 notificaciones por cada 100.000 dosis administradas.
Estas notificaciones españolas se comparten con las del resto de las 27 agencias europeas y con la OMS, al enviarse a la base de datos europea (EudraVigilance) y a la base de datos de la OMS (VigiBase), respectivamente. Según estas cifras, España es el sexto país de la Unión Europea que más notificaciones de acontecimientos adversos tras la administración de vacunas frente a COVID-19 ha enviado a la base de datos EudraVigilance.
Las vacunas frente a la COVID-19 se han estudiado antes de su comercialización en un número de sujetos muy elevado, entre 30.000 y 50.000 voluntarios, y en el momento de su utilización se dispone de información de varios meses de seguimiento. Todo esto permite conocer las reacciones adversas frecuentes que se presentan en los primeros meses, periodo en el que normalmente se presentan en la mayoría de las RAM de las vacunas, mejor identificadas como ESAVI.
Con todo lo descrito anteriormente, podemos decir que el éxito de la campaña de farmacovigilancia de las vacunas frente a las COVID-19 nos hace seguir recomendando la utilización del Sistema Español de Farmacovigilancia Humana (SEFV-H) como una herramienta fundamental para el uso racional de los medicamentos. Solo conociendo las posibles reacciones adversas a los medicamentos o los acontecimientos adversos relacionados con las vacunas podemos seguir utilizando de manera adecuada estas herramientas terapéuticas.
Por todo ello, se agradece a todos los profesionales sanitarios y a los ciudadanos el esfuerzo realizado en 2021 para la notificación de las sospechas de reacciones adversas, en particular con las vacunas frente a COVID-19. Su colaboración ha sido, y será, esencial para mejorar el conocimiento del perfil en los medicamentos. Por lo tanto solo resta seguir solicitando la notificación de todas las sospechas de reacciones adversas de los medicamentos, en particular las de los de reciente autorización (triángulo▼) por los medios establecidos en el Sistema Español de Farmacovigilancia Humana (ver Información importante, al final de esta sección).
La agencia reguladora de medicamentos de Japón (PMDA) ha informado del riesgo de lesiones óseas graves en el recién nacido, si se ha utilizado el sulfato de magnesio, en casos de eclampsia, durante mucho tiempo en el embarazo.
En Japón, desde su Ministerio de Salud (MHLW) y desde su Agencia Reguladora de Medicamentos (Pharmaceutilcals and Medical Devices Agency, PMDA) han comunicado que la información del medicamento con el sulfato de magnesio (inyectable), indicado para la eclampsia, debe revisarse para incluir el riesgo de lesiones óseas similares al raquitismo en los recién nacidos al nacer, asociados con la administración prolongada de este medicamento durante el embarazo (PMDA, 2021).
El MHLW y el PMDA revisaron casos de lesiones óseas similares al raquitismo notificados en recién nacidos de mujeres pacientes tratadas con sulfato de magnesio en Japón. Y concluyeron que una relación causal entre el medicamento y el evento era razonablemente posible en todos los casos. La duración más corta de la administración con sulfato de magnesio (inyecciones) a la madre fue de 18 días.
En España, en la información de la ficha técnica de los medicamentos con sulfato de magnesio inyectable (Sulmetin Simple® y Sulfato de Magnesio Altan® EFG) en presentaciones de 150 mg/ml de solución inyectable y para perfusión, en la sección 6. Fertilidad, embarazo y lactancia se describe lo siguiente:
Embarazo: El sulfato de magnesio puede causar anomalías fetales cuando se administra más de 5-7 días para las mujeres embarazadas.
Hay estudios epidemiológicos retrospectivos y casos clínicos que documentan anomalías fetales como la hipocalcemia, y alteraciones esqueléticas de desmineralización. Cuando se administra por perfusión intravenosa (no debe administrarse durante las 2 horas previas al parto) en toxemia del embarazo, el recién nacido puede mostrar signos de toxicidad por magnesio, incluyendo depresión neuromuscular o depresión respiratoria.
La utilización de magnesio durante el embarazo sólo debe plantearse en el caso de que sea absolutamente necesario. En estos casos se recomienda monitorizar los niveles plasmáticos de magnesio, la presión arterial, frecuencia respiratoria y reflejos tendinosos profundos.
El profesional sanitario debe conocer este riesgo de su uso durante el embarazo, y debe seguir las indicaciones de la ficha técnica con el resumen de las características farmacoterapéuticas del medicamento.
La utilización del sulfato de magnesio puede causar anomalías fetales, cuando se administra más de 5-7 días para las mujeres embarazadas.