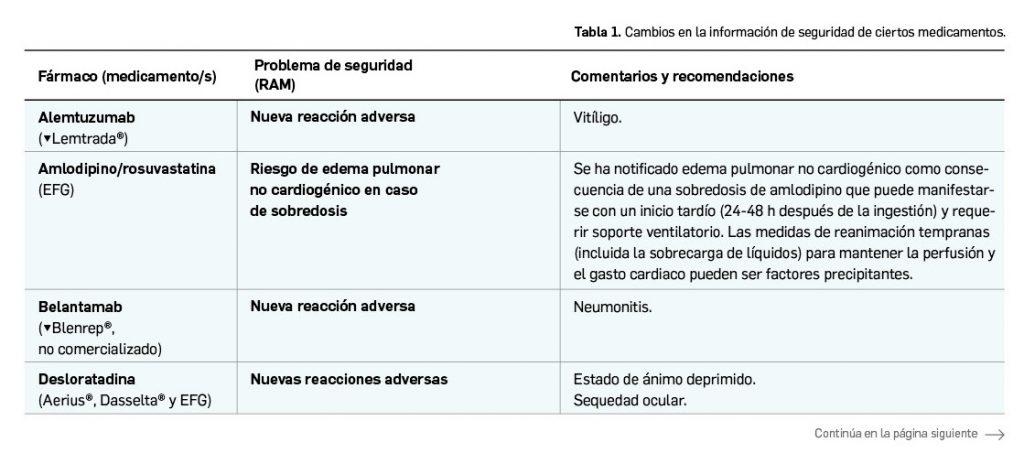
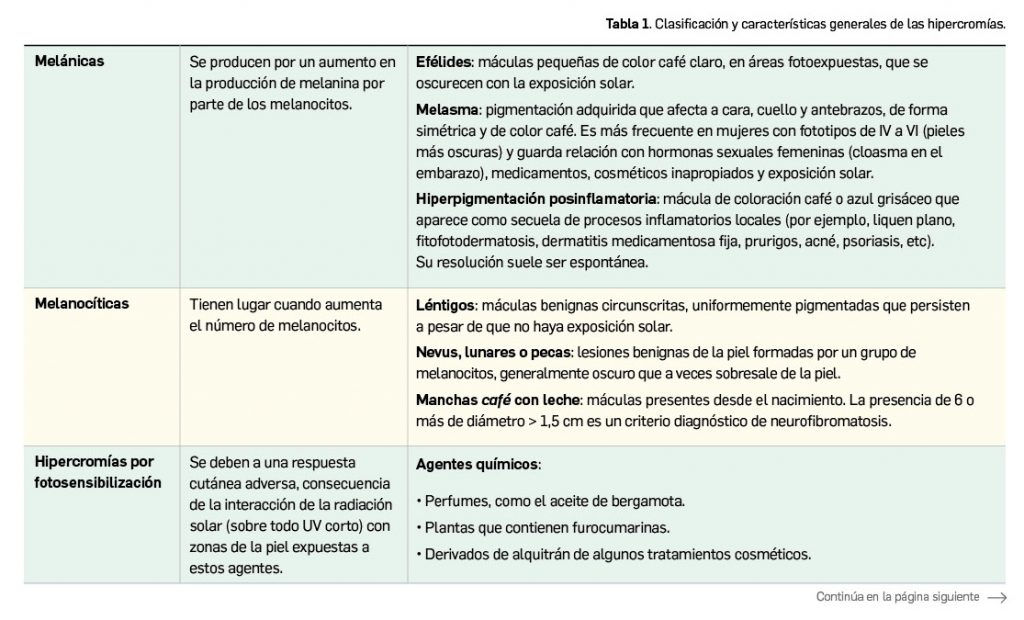
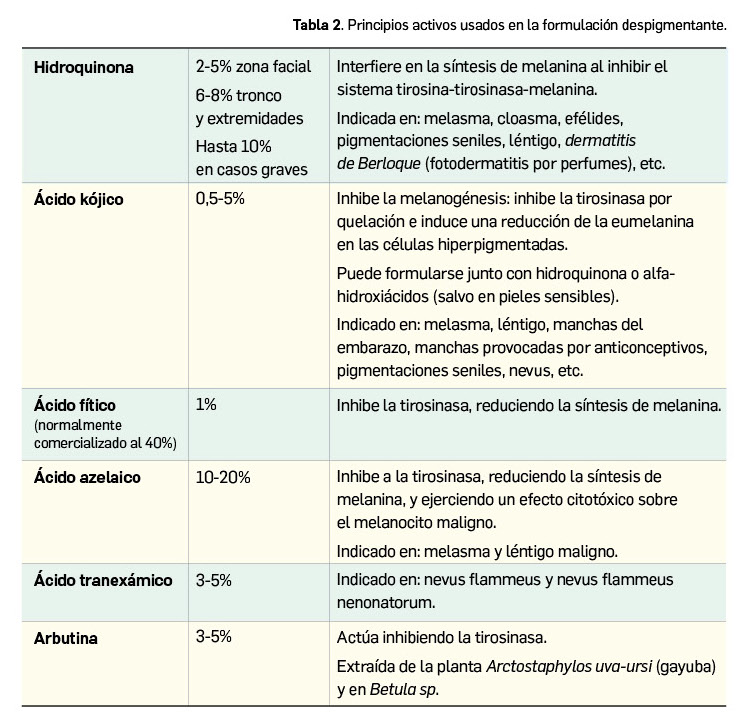
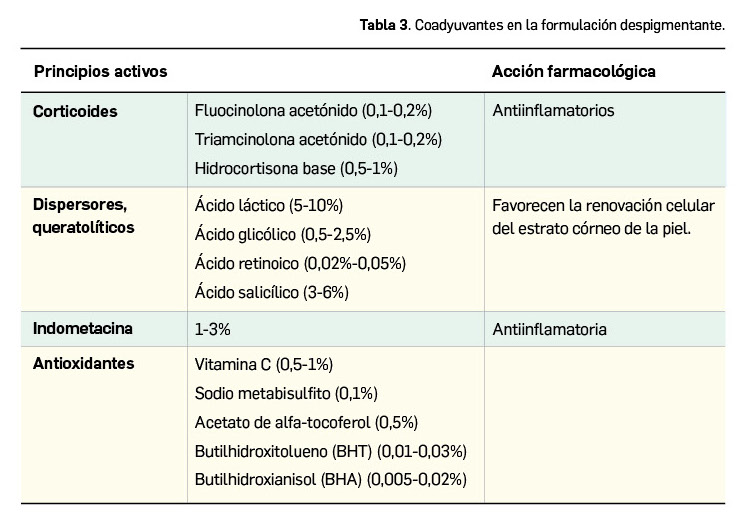
La agencia de medicamentos de Australia ha informado del riesgo potencial de hipertensión arterial en pacientes tratados con erenumab, un fármaco relativamente nuevo indicado en la profilaxis de la migraña en adultos con al menos 4 días de migraña al mes.
La autoridad reguladora de Australia, la Therapeutic Goods Administration (TGA), ha anunciado que la información del medicamento con erenumab (▼Aimovig®) se ha actualizado con la inclusión de una advertencia sobre una posible relación causal entre el fármaco y la hipertensión (TGA, 2021).
Erenumab está indicado para la profilaxis de la migraña en adultos con al menos 4 días de migraña al mes.
La TGA ha revisado casos de desarrollo de hipertensión arterial y de empeoramiento de la hipertensión preexistente después uso del fármaco en el periodo posautorización a nivel internacional. Se ha concuido que la hipertensión puede ocurrir en cualquier momento durante el tratamiento, pero ha sido notificada con más frecuencia dentro de los siete días posteriores a la administración de una dosis; en la mayoría de los casos, la aparición o empeoramiento de la hipertensión se comunicó después de la primera dosis del medicamento.
Recomendaciones
Los profesionales sanitarios deben monitorear a los pacientes tratados con erenumab (▼Aimovig®) debido a posibles casos de nueva aparición de hipertensión o de empeoramiento de la hipertensión preexistente. Si se observa hipertensión y la evaluación no logra establecer una etiología alternativa, debe valorarse la posible interrupción del tratamiento con erenumab.