Número 454, Junio 2022
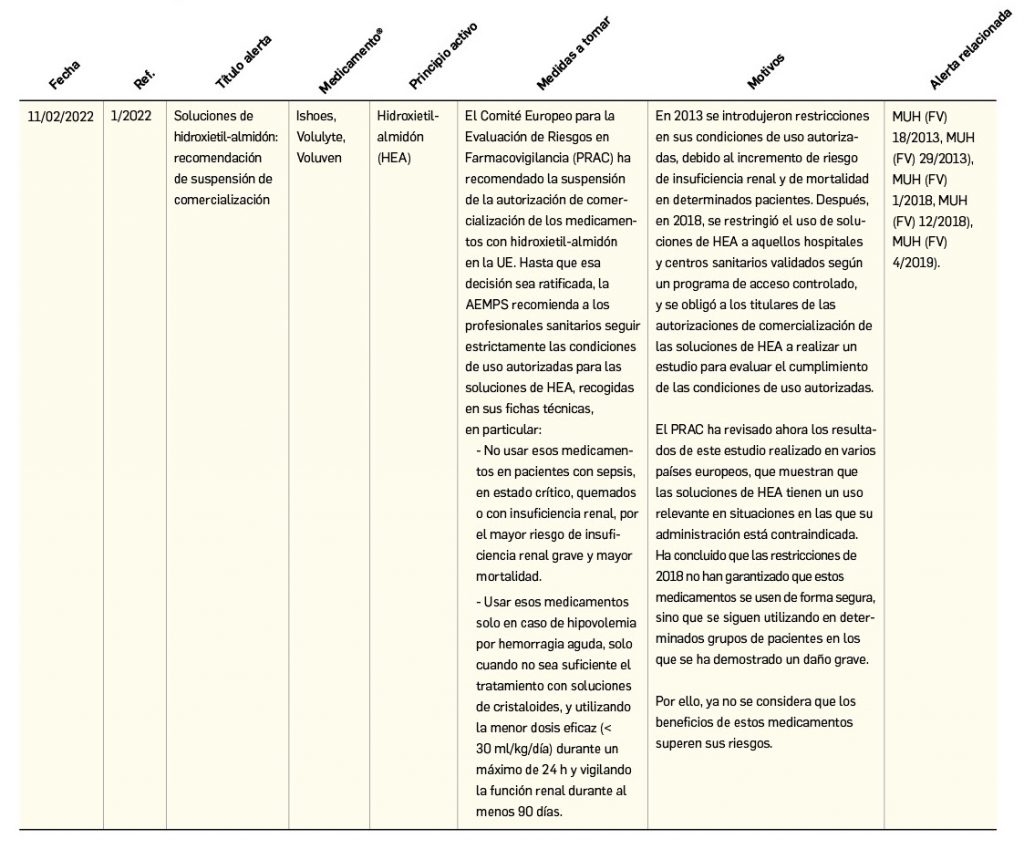
Resumen de las notas sobre seguridad y farmacovigilancia publicadas por la AEMPS desde principios del año 2022. Para información más ampliada y acceso al documento de la AEMPS, puede consultar BOT PLUS.
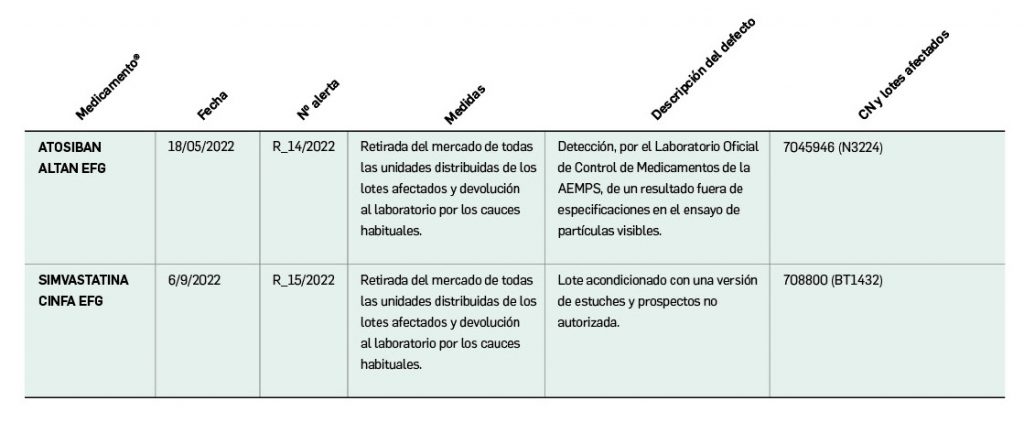
Alertas debidas a defectos de calidad observados en medicamentos de uso humano, publicadas por la AEMPS desde el anterior número y que suponen la retirada o inmovilización de ciertos lotes de medicamentos. En BOT PLUS puede encontrar más información detallada, con acceso al documento de la AEMPS.
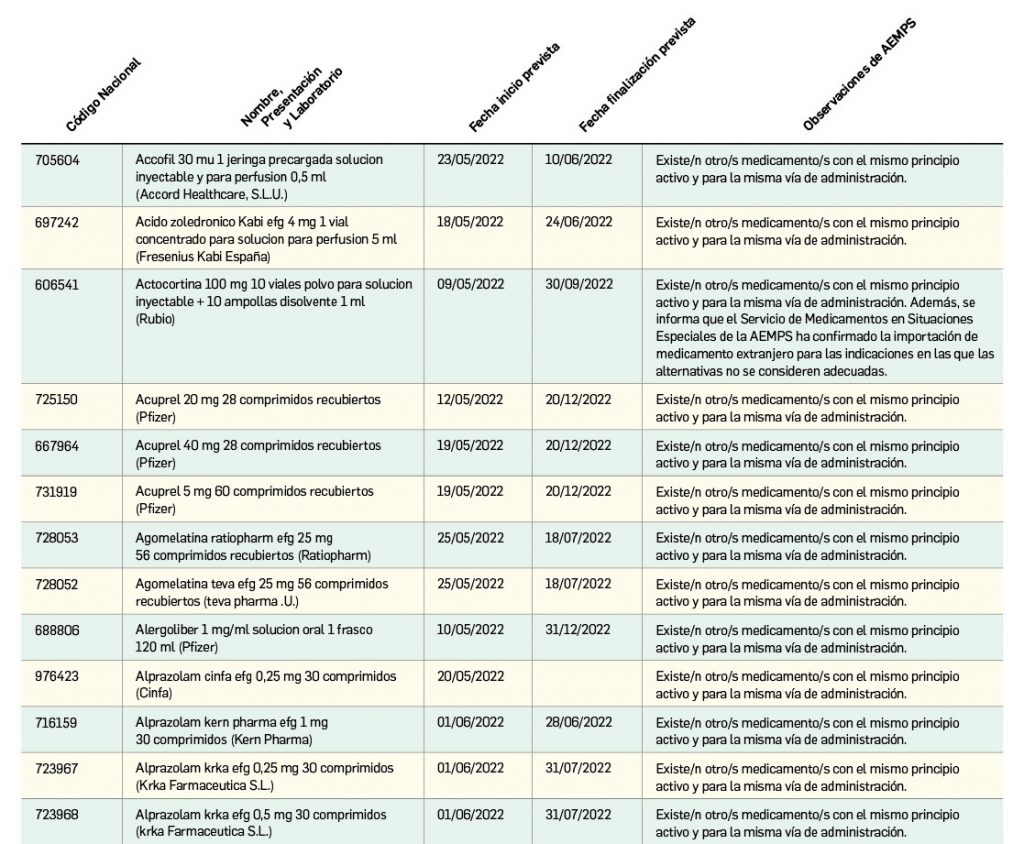
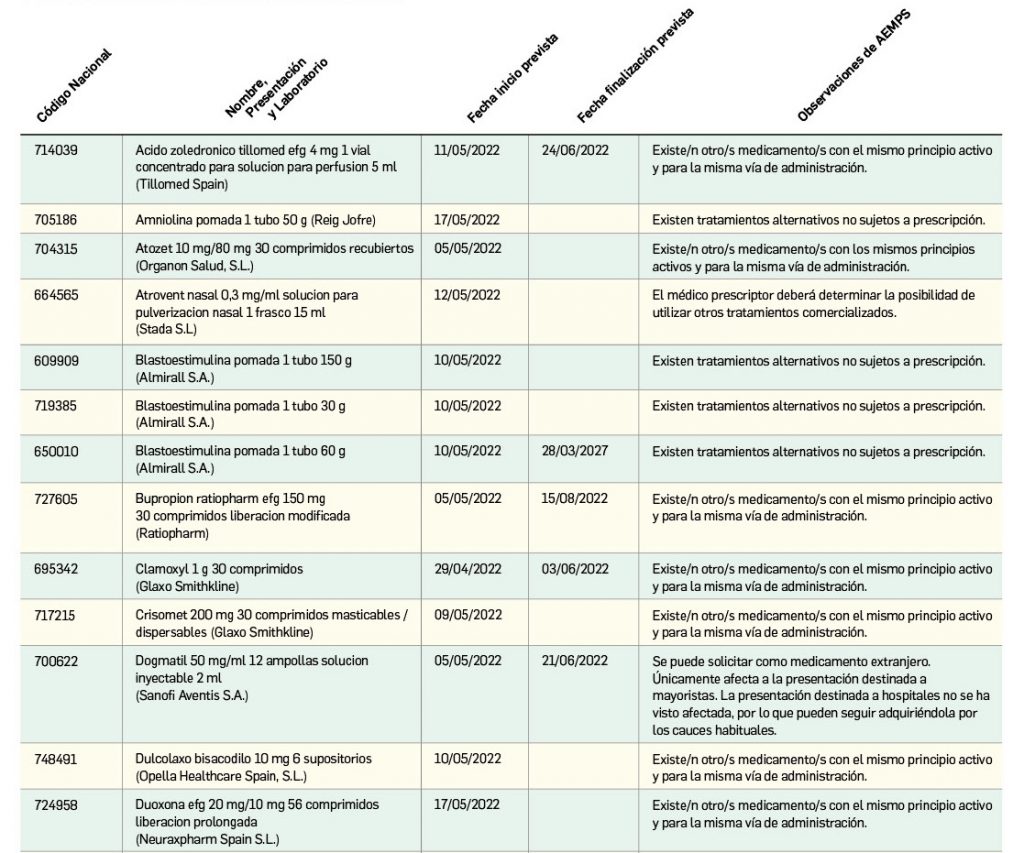
Listado de medicamentos con problemas de suministro publicado por la AEMPS, a fecha de cierre de este número. En BOT PLUS, se puede encontrar la información completamente actualizada, al tratarse de una información que varía de forma continua.
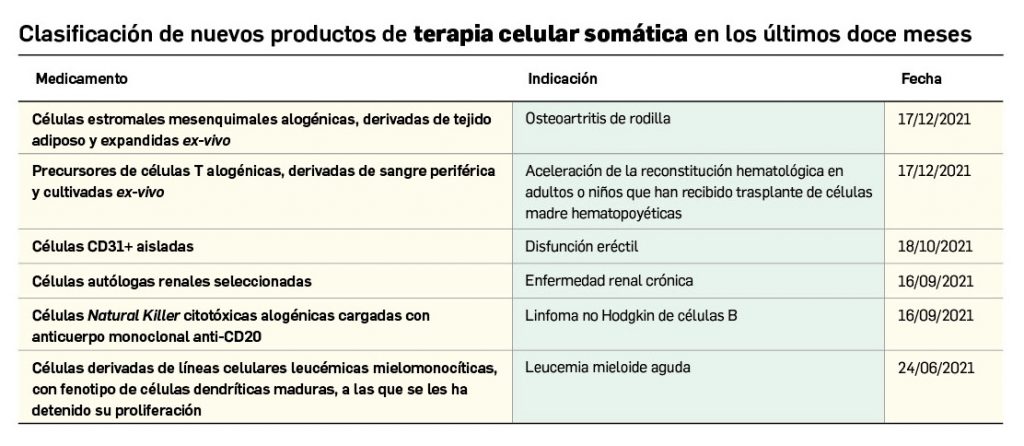
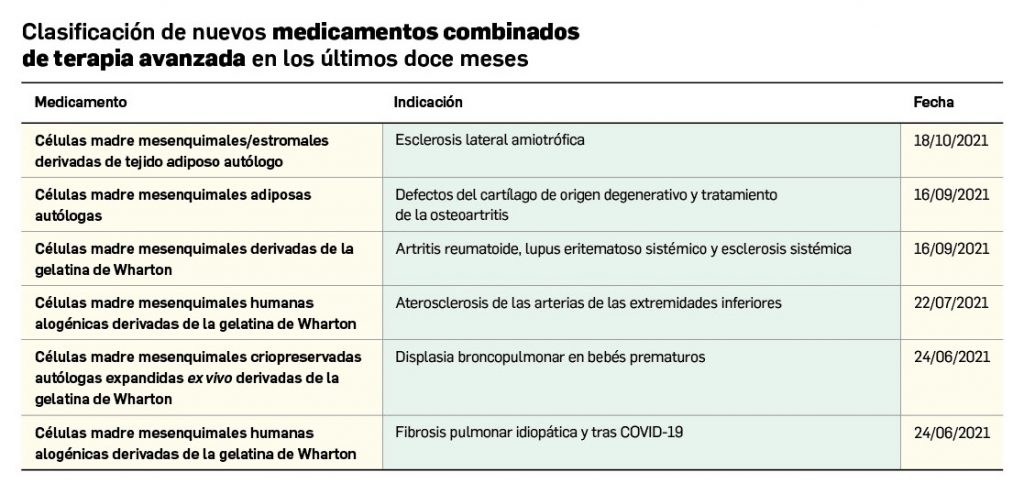
Los medicamentos de terapia avanzada (MTA o Advanced Therapy Medicinal Products, ATMP) ofrecen nuevos e innovadores tratamientos para las enfermedades. Están basados en la terapia génica, la terapia celular somática o la ingeniería tisular. El marco legal para las ATMP en la Unión Europea está establecido en la Regulation (EC) No 1394/2007 on advanced therapy medicinal products que asegura el libre movimiento de estas medicinas dentro de la Unión Europea y el acceso a los mercados. La regulación (EC) nº 1394/2007 también establece el nuevo Comité en Terapias avanzadas (CAT) cuya responsabilidad fundamental consiste en preparar un proyecto de opinión sobre cada nueva solicitud de medicamento de terapia avanzada planteada a la Agencia Europea de Medicamentos, antes de que el Comité de Medicamentos de Uso Humano (CHMP, Committee for Medicinal Products for Human Use) de la misma adopte una opinión definitiva sobre la concesión, modificación, suspensión o revocación de una autorización de comercialización para el medicamento en cuestión.


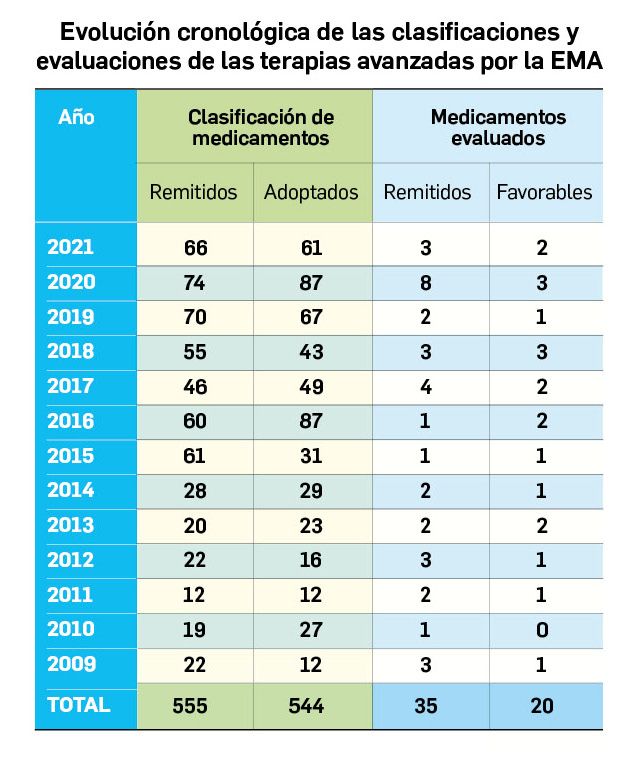
Los medicamentos huérfanos son aquéllos que sirven para diagnosticar, prevenir o tratar enfermedades raras de carácter muy grave o con riesgo para la vida. En la Unión Europea, la calificación de enfermedad rara se aplica a todas aquellas que no afectan a más de 5 de cada 10.000 personas. La designación de un medicamento como huérfano no garantiza su uso en la condición designada y no implica necesariamente que el producto satisfaga los criterios de eficacia, seguridad y calidad necesarios para la concesión de la autorización de comercialización. Como para cualquier medicamento, estos criterios solo pueden ser evaluados una vez que la solicitud de autorización de comercialización haya sido presentada.

► Instituto de Salud Carlos III (Ministerio de Ciencia e Innovación):
Instituto de Investigación en Enfermedades Raras:
https://www.isciii.es/QuienesSomos/CentrosPropios/IIER/Paginas/default.aspx
CIBERER (Centro de Investigación Biomédica en Red de Enfermedades Raras): https://www.ciberer.es/
► Instituto de Mayores y Servicios Sociales (IMSERSO, Ministerio de Derechos Sociales y Agenda 2030):
http://www.imserso.es/imserso_01/index.htm
► Federación Española de Enfermedades Raras (FEDER): www.enfermedades-raras.org
► Asociaciones de pacientes en España: https://enfermedades-raras.org/index.php/asociaciones/nuestros-socios
► Agencia Europea de Medicamentos (EMA; Europea Medicines Agency). Apartado de Medicamentos Huérfanos
(inglés): https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview
https://www.ema.europa.eu/en/committees/committee-orphan-medicinal-products-comp
► Comisión Europea: web oficial de la Comisión Europea sobre enfermedades raras y medicamentos huérfanos (español).
http://ec.europa.eu/health/rare_diseases/policy/index_es.htm
► Orphanet: Portal de información oficial de la Unión Europea sobre enfermedades raras y medicamentos huérfanos (español).
http://www.orpha.net/consor/cgi-bin/index.php?lng=ES
► Eurordis: Federación Europea de Asociaciones de Pacientes con Enfermedades Raras (español). http://www.eurordis.org/es
► Food & Drug Administration (FDA, Estados Unidos). Apartado de Medicamentos Huérfanos (inglés):
http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/default.htm
► Pharmaceuticals & Medical Devices Agency. Agencia de Medicamentos y Dispositivos Médicos, de Japón (inglés):
http://www.pmda.go.jp/english/index.html
La agencia de medicamentos del Reino Unido (MHRA) ha informado del riesgo de hipercalcemia grave, y potencialmente mortal, con denosumab 60 mg (Prolia®) cuando se usó en niños y adolescentes en ensayos clínicos para la osteogénesis imperfecta y durante el uso no indicado en la ficha técnica autorizada.
La autoridad reguladora del Reino Unido, MHRA, ha informado sobre los resultados de una revisión europea reciente, en la que se evaluaron casos de hipercalcemia grave y ha recomendado advertencias más estrictas contra el uso de Prolia® en niños y adolescentes menores de 18 años. Se ha considerado esta revisión junto con los datos de seguridad y se ha acordado que la información del producto debe actualizarse (MHRA, 2022).
Dichas acciones se han debido a la notificación de casos de hipercalcemia grave y potencialmente mortal que requirieron hospitalización y se complicaron con una lesión renal aguda, en niños y adolescentes menores de 18 años que recibieron 60 mg de denosumab (Prolia®) en ensayos clínicos. Estos ensayos clínicos investigaban el tratamiento con denosumab en pacientes menores de 18 años para el abordaje de la osteogénesis imperfecta, que consiste en un grupo de condiciones hereditarias raras que provocan que los huesos sean muy frágiles.
En todo el mundo, se ha reunido información de 20 notificaciones de sospechas de reacción adversa a medicamentos (RAM) de hipercalcemia comunicados hasta el 26 de agosto de 2021, durante el tratamiento no autorizado con Prolia® en niños y adolescentes menores de 18 años. Las notificaciones incluyeron casos en pacientes pediátricos con osteogénesis imperfecta, así como en aquellos con otras patologías. También hubo un pequeño número de notificaciones de hipercalcemia en pacientes menores de 18 años después de suspender el tratamiento (hipercalcemia de rebote).
Los síntomas de la hipercalcemia incluyen: sed excesiva, micción excesiva, somnolencia, confusión, pérdida de concentración, náuseas o vómitos, estreñimiento y debilidad muscular. La hipercalcemia grave puede causar problemas renales graves (lesión renal aguda), coma, alteraciones del ritmo cardiaco y paro cardiaco.
En consecuencia, la ficha técnica o resumen de las características del producto (SmPC, por sus siglas en inglés) de Prolia® se va a actualizar para recomendar que denosumab 60 mg no se debe usar en niños y adolescentes menores de 18 años debido a problemas de seguridad relacionados con la hipercalcemia grave. También existen advertencias de que la inhibición de RANK/ligando RANK (RANKL) en estudios con animales puede estar asociada con la inhibición del crecimiento óseo y la falta de erupción dental.
Se recuerda a los profesionales sanitarios lo siguiente:
Los profesionales sanitarios deben de informar a los pacientes o a los padres y cuidadores sobre estos aspectos:
La Agencia Europea de Medicamentos ha publicado nueva información a partir del seguimiento permanente de las RAM notificadas con el uso de las vacunas frente a la COVID-19. Respecto de las vacunas de ARNm, se ha evaluado la evidencia sobre su asociación con la aparición de hepatitis autoinmune, y se ha concluido que, en el momento actual, no hay evidencia suficiente para establecer su asociación con la aparición de hepatitis autoinmune.
La Agencia Europea de Medicamentos (EMA) ha emitido nueva información a partir del seguimiento permanente de las RAM notificadas con el uso de las vacunas frente a la COVID-19 (AEMPS, 2022).
En relación con las vacunas de ARNm –▼Comirnaty® (Pfizer-BioNTech) y ▼Spikevax® (Moderna)– recuerda que, desde su autorización de comercialización en la UE el 21 de diciembre de 2020 y hasta el 24 de abril de 2022, se han administrado en el Espacio Económico Europeo (EEE) alrededor de 600 millones de dosis de Comirnaty® en adultos y alrededor de 27 millones de dosis en niños y adolescentes menores de 18 años; y desde el 6 de enero de 2021 (fecha de la comercialización en la UE) hasta el 24 de abril de 2022 se han administrado alrededor de 153 millones de dosis de Spikevax® en adultos y alrededor de 1,9 millones de dosis en niños y adolescentes menores de 18 años.
Respecto a la hepatitis autoinmune se puede describir como una enfermedad inflamatoria crónica y grave, en la que el sistema inmune ataca y daña el hígado. Sus signos y síntomas varían entre las personas y pueden incluir coloración amarilla de la piel (ictericia), acumulación de líquidos en las piernas (edema), o síntomas gastrointestinales (ascitis).
La revisión europea se ha llevado a cabo en base a la información procedente de la literatura científica, los casos de hepatitis autoinmune notificados a nivel mundial y los datos adicionales que han proporcionado los titulares de autorización de comercialización. Así, el comité europeo de evaluación de riesgos en farmacovigilancia (PRAC, por sus siglas en inglés) ha concluido que la evidencia disponible hasta el momento no apoya la existencia de una relación causal entre la administración de Comirnaty® y Spikevax® y los casos de hepatitis autoinmune notificados. No obstante, este riesgo continuará bajo estrecha vigilancia y se tomarán las medidas apropiadas en caso necesario.
Como recomendaciones actuales en relación con hepatitis autoinmune, aspecto revisado por la EMA con las dos vacunas de ARNm, se informa de que: