Resumen
Isatuximab es un nuevo anticuerpo monoclonal que se une específicamente al receptor CD38, el cual se expresa uniforme e intensamente por las células del mieloma múltiple (MM). Esa unión media la activación de diversos mecanismos citotóxicos e inmunitarios que acaban con la lisis o apoptosis de las células mielomatosas y permiten controlar el crecimiento tumoral. El medicamento ha sido autorizado, en combinación con pomalidomida y dexametasona, para el tratamiento de pacientes adultos con MM resistente al tratamiento o recidivante que han recibido ≥ 2 tratamientos previos, incluyendo lenalidomida y un inhibidor del proteasoma y han mostrado progresión de la enfermedad tras el último tratamiento; y en combinación con carfilzomib y dexametasona para el tratamiento de pacientes adultos con MM que han recibido al menos un tratamiento previo.
Su aprobación se ha sustentado en dos ensayos clínicos de diseño adecuado, que han confirmado su favorable balance beneficio-riesgo. El primero demostró la superioridad de la adición de isatuximab a la combinación de pomalidomida y dexametasona, en comparación con el solo uso de estos dos fármacos como control activo, en pacientes con MM en progresión tras haber recibido ≥ 2 líneas previas, incluyendo lenalidomida y un inhibidor de proteasoma: prolongó la mediana de SLP en unos 5 meses (11,5 vs. 6,5 meses), reduciendo el riesgo de progresión de la enfermedad o muerte en un 40%. También mejoró la tasa de respuesta antitumoral (60% vs. 35%) y su profundidad, resultando en una tendencia favorable en SG –prolongación de la mediana en casi 7 meses (24,6 vs. 17,7)– que aún debe confirmarse con los análisis finales. El segundo estudio evidenció que la asociación del nuevo fármaco a la combinación de carfilzomib y dexametasona mejora los resultados de la doble terapia con estos dos últimos en pacientes con MM en recaída que habían recibido entre 1 y 3 líneas previas de tratamiento (excluyendo carfilzomib y daratumumab): prologó notablemente la SLP (mediana no alcanzada en el brazo experimental vs. 19,1 meses en el control), reduciendo en un 47% el riesgo de progresión o muerte. Sin datos maduros de SG, la tasa y profundidad de la respuesta también fueron mayores con isatuximab (muy buena respuesta parcial o mejor en 73% vs. 56%). La eficacia del nuevo fármaco se reveló rápida, duradera y consistente en los distintos subgrupos de pacientes evaluados, con independencia de la refractariedad a fármacos concretos.
No obstante, su perfil toxicológico es importante y se asocia a una alta frecuencia de reacciones adversas, graves en casi 2 de cada 3 pacientes. Los eventos adversos de mayor incidencia, en su mayoría manejables en clínica, serían esperables por tratarse de una proteína inmunomoduladora y a la vista de la seguridad de los fármacos con que se combina. Sobresalen por su frecuencia (≥ 20%) las infecciones del tracto respiratorio superior e inferior (incluidas bronquitis y neumonía), citopenias (sobre todo, neutropenia), reacciones a la perfusión, alteraciones del tracto gastrointestinal (náuseas y diarrea) y otras inespecíficas, como fatiga o insomnio. Por su gravedad, las infecciones y, en particular, la neumonía, parecen constituir el mayor riesgo asociado al fármaco.
Isatuximab no aporta ninguna novedad en cuanto a mecanismo de acción: comparte vía terapéutica con el ya comercializado daratumumab. Las comparaciones indirectas disponibles entre ambos sugieren que no supera las mejoras incorporadas por daratumumab, el cual tiene un espectro de indicaciones más amplio (incluso en 1ª línea) y se considera de elección en esquemas de rescate. En definitiva, el nuevo fármaco representa una alternativa más de triple terapia a partir de la 2ª línea de tratamiento, y su uso puede ser especialmente relevante en pacientes refractarios a lenalidomida; en ese contexto, se suma a otros fármacos biológicos previamente disponibles (por ejemplo, daratumumab o elotuzumab) sin aportar ningún elemento de innovación terapéutica disruptiva. Además, la futura disponibilidad de las prometedoras terapias CAR-T (idecabtagén vicleucel) hace previsible un cambio en los enfoques de tratamiento.
Aspectos fisiopatológicos
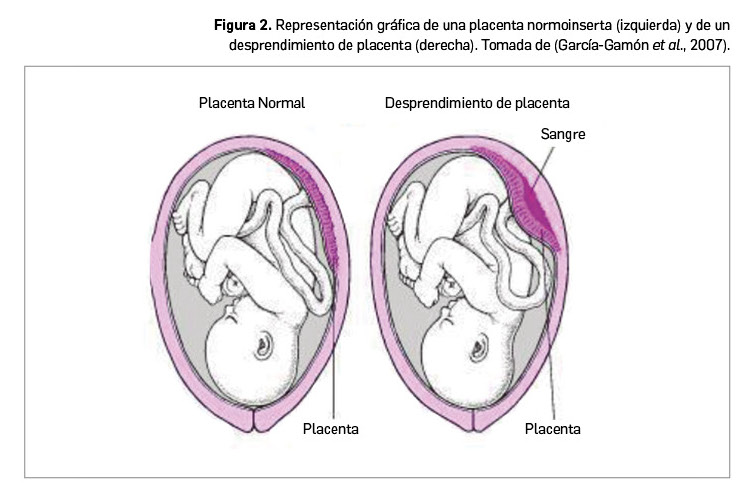
El mieloma múltiple (MM) o mieloma de células plasmáticas es un agresivo tumor hematológico maligno de células o linfocitos B, caracterizado por la proliferación en la médula ósea de un clon de células plasmáticas (procedentes de la maduración de células B) que, generalmente, producen y secretan una paraproteína monoclonal detectable en suero o en orina como signo subyacente al daño orgánico. Presenta algunas características similares a la leucemia y supone aproximadamente el 1% de los casos de cáncer y, específicamente, el 10% de los tumores hematológicos. No obstante, dado su actual carácter de incurable, representa el 2% de la mortalidad por cáncer y el 20% de la de los cánceres hematológicos.
La incidencia en la UE es de 4,5-6,0 casos por cada 100.000 habitantes/año, lo que se traduce en una cifra de en torno a 40.000 nuevos casos anuales; sin embargo, dado que la mortalidad es elevada (4,1 muertes/100.000/año, con una supervivencia a 5 años en torno al 47%), la prevalencia no es muy alta. En general, los países del sur de Europa –España incluida– tienen tasas de incidencia y mortalidad más bajas que los del norte, pero tanto en unos como está en aumento. En España se registran entre 1.500 y 2.000 casos al año (40 nuevos casos por cada millón de habitantes). La mediana de edad de diagnóstico se sitúa en torno a los 65-70 años y solo un 2% de los casos diagnosticados corresponden a pacientes menores de 40 años. Así, la incidencia de MM en España es de 1,5-2,5 casos nuevos/100.000 en menores de 65 años, pero sube a 25-30 a partir de esa edad, afectando a un número ligeramente superior de hombres que de mujeres (ratio 1,4:1).
El MM forma parte de las denominadas gammapatías monoclonales, un grupo heterogéneo de enfermedades caracterizadas por una producción anormal de inmunoglobulinas (Ig) y la aparición de tumores de células plasmáticas. Las células plasmáticas malignas características del MM –células mielomatosas– se acumulan en la médula ósea y pueden llegar al torrente sanguíneo, provocando una alteración de la función normal de la médula, daños óseos y alteración de la función inmune. Estas células pueden también formar tumores localizados, los plasmocitomas, que pueden tener una localización tanto ósea como extraósea: es cuando coexisten varios plasmocitomas con diversas localizaciones cuando se emplea apropiadamente el término mieloma múltiple.
La característica típica de la célula mielomatosa es, pues, la producción y liberación al torrente circulatorio de una Ig monoclonal, denominada proteína M, pero también conocida como proteína mielomatosa, para-proteína o proteína en pico (así llamada por su identificación como una banda en forma de pico en el proteinograma por electroforesis). Se trata de una Ig producida como consecuencia de la aparición de una o más mutaciones en los genes responsables de la producción de Ig en la célula mielomatosa. Tiene una secuencia de aminoácidos y una estructura anormales en relación a las inmunoglobulinas fisiológicas, que provocan la adherencia entre las moléculas y con las estructuras celulares y tisulares tejidos: células sanguíneas, pared de los vasos sanguíneos u otros componentes de la sangre. Todo ello provoca la disminución del flujo sanguíneo, causando un síndrome
de hiperviscosidad.
En un 30% de los casos se produce una mayor cantidad de cadenas ligeras de la requerida para combinarse con las cadenas pesadas de las inmunoglobulinas, dando lugar a la proteína de Bence-Jones, la cual tiene un peso molecular de 22,000 Da y es lo suficientemente pequeña como para ser excretada por orina y producir un aumento de nivel de proteínas urinarias. Estas proteínas también pueden adherirse entre sí o con otros tejidos, dando lugar a amiloidosis –depósitos proteicos en cualquier tejido del organismo, como riñón, tejido nervioso o músculo cardiaco– o a la enfermedad de depósito de cadenas livianas (dichas cadenas se depositan al azar, pero especialmente en los pequeños vasos del ojo o del riñón). Las proteínas monoclonales anormales también pueden provocar un amplio abanico de perturbaciones fisiológicas, al unirse a los factores de la coagulación (trastornos diversos de coagulabilidad) y a otras sustancias circulantes, con diversas consecuencias hormonales y metabólicas.
La Beta 2 microglobulina (β2M) es otra pequeña proteína cuyos niveles se encuentran elevados en pacientes con MM activo, si bien un 10% de los pacientes no la producen. Precisamente, el estadiaje internacional del MM se basa en las concentraciones en suero de albúmina y de microglobulina β2: el estadio I se caracteriza por niveles de β2M inferiores a 3,5 mg/l y de albúmina mayores de 3,5 g/l; el estadio II por β2M < 3,5 mg/l y de albúmina < 3,5 g/, o bien β2M de 3,5 a 5,5 mg/l; finalmente, el estadio III se caracteriza por β2M > 5,5 mg/l. Estos estadios se asocian con medianas de supervivencia progresivamente inferiores: 62, 44 y 29 meses, respectivamente.
La etiología del MM, como para la práctica totalidad de tumores, se considera multifactorial. No se conocen con exactitud las causas directas, y aunque no se ha detectado un factor genético concreto que determine el desarrollo de la patología, suelen predominar eventos oncogenéticos primarios o secundarios que originan una proliferación descontrolada de un clon celular neoplásico; por ejemplo, se ha descrito que las traslocaciones entre el cromosoma 14q32 y sus vecinos, y la desregulación del oncogén c-myc parecen jugar un papel clave en las etapas iniciales de la enfermedad. De igual modo, se ha asociado el MM con la exposición a radiaciones ionizantes, y se ha propuesto un mayor riesgo asociado a exposiciones ocupacionales –a sustancias químicas, como benceno, asbesto, pesticidas o pinturas y disolventes– relacionadas con la agricultura, refinerías, industrias del corcho, del metal, del plástico o de la madera, o si se ha trabajado como conductor de camiones. También se ha sugerido la posible relación con algunas enfermedades autoinmunes, como la artritis reumatoide.
Sea como fuere, el crecimiento descontrolado de las células mielomatosas tiene importantes consecuencias clínicas: la destrucción del esqueleto, la insuficiencia de la médula ósea, hipervolemia e hiperviscosidad sanguíneas, supresión de la producción de inmunoglobulinas normales e insuficiencia renal. Pese a que el MM puede permanecer asintomático durante muchos años, en la fase sintomática el dolor óseo es el cuadro de presentación más común; las células mielomatosas y el aumento del número de osteoclastos parecen ser las responsables de la destrucción ósea.
Fisiopatología ósea en el mieloma múltiple
El mecanismo que produce la activación de los osteoclastos es complejo, pero se ha observado que participan diversas citocinas locales, tales como interleucinas (IL-1b, IL-6), quimiocinas (por ejemplo, las MIP-1) y las integrinas implicadas en el proceso de adhesión celular. Entre las citocinas con un papel más relevante, quizá la mejor caracterizada sea la IL-6: producida fundamentalmente por las células madre de la médula ósea y los macrófagos, es un importante mediador del crecimiento, supervivencia y migración de las células del MM, e incluso de la resistencia a la quimioterapia, y actúa como un estímulo paracrino de las células plasmáticas. Por otro lado, la adhesión de las células plasmáticas al estroma de la médula ósea incrementa la secreción por este de IL-6, completando un auténtico sistema de retroalimentación que potencia la tumorigénesis plasmática; en definitiva, IL-6 parece tener un papel fundamental en la patología del MM, potenciando la supervivencia de las células plasmáticas e inhibiendo su apoptosis. El efecto acumulativo de este complejo ambiente bioquímico da lugar a un desequilibrio de las fuerzas anti- y proapoptóticas dentro de las células de mieloma, favoreciendo la expansión clonal desregulada y la proliferación tumoral.
Actualmente, se considera que los factores efectores finales que regulan la remodelación ósea forman parte de la superfamilia del Factor de Necrosis Tumoral (TNF) y de la de su receptor; entre ellos puede citarse el RANKL (ligando del receptor del activador del factor nuclear kappa-B), cuya producción es máxima en las células indiferenciadas del estroma osteoclástico y se reduce a medida que madura el fenotipo osteoblástico. Estimula la diferenciación, supervivencia y fusión de las células precursoras de osteoclastos, activa los osteoclastos maduros y prolonga su vida útil. Como resultado, permite la expansión de la masa osteoclástica activa capaz de formar sitios de resorción ósea.
El tejido nervioso se ve también afectado con frecuencia en los pacientes con mieloma, tanto por los efectos directos de los anticuerpos de las proteínas mielomatosas frente a la mielina como por el depósito de fibrillas proteicas (amiloide), que resultan en neuropatías periféricas. Tanto en el hueso como en los tejidos blandos puede producirse una compresión o desplazamiento de los nervios procedentes de la médula espinal o del tallo cerebral. Además, por la susceptibilidad a las infecciones, las infecciones virales de los tejidos nerviosos son muy frecuentes, en especial por varicela zóster y parálisis de Bell.
La predisposición a las infecciones es probablemente el rasgo más característico de los pacientes con mieloma junto con la enfermedad ósea, debido a la inhibición de las funciones inmunitarias normales: producción deficitaria de anticuerpos normales, daño de la función de los linfocitos T y activación anómala de la función monocito/macrófago. Los pacientes con MM son particularmente susceptibles a las infecciones virales y a las infecciones con bacterias encapsuladas, como el neumococo.
Terapéutica
El tratamiento se basa fundamentalmente en la supresión de las células mielomatosas mediante quimio y/o radioterapia, asociado eventualmente a trasplante de células progenitoras hematopoyéticas periféricas.
El fármaco antineoplásico más comúnmente utilizado ha sido melfalán, clásicamente considerado el mejor agente único para el tratamiento del MM. La mayoría de los pacientes presentan respuesta a este fármaco, particularmente cuando se utiliza combinado con prednisona. De hecho, la asociación melfalán/prednisona (protocolo MP) se usa con frecuencia, dando lugar a respuestas objetivas en el 60% de los pacientes; estas respuestas se manifiestan normalmente con un 50% de mejora en los niveles de proteína M, en los recuentos sanguíneos y en otros resultados bioquímicos, además de la mejoría de varios síntomas de la enfermedad, como el dolor óseo y la fatiga. La ciclofosfamida puede reemplazar a melfalán, con una actividad antimielomatosa similar. Asimismo, los resultados alcanzados con bendamustina son equiparables a los obtenidos con melfalán; incluso, la tasa de respuestas completas es sustancialmente mayor y el porcentaje de pacientes supervivientes a los 5 años también, aunque en ambos casos es una tasa moderada (29% vs. 19%). Pese a que se han desarrollado combinaciones quimioterápicas más complejas con respuestas similares o ligeramente mejores, no está claro que sean de primera elección.
Diferentes líneas de investigación facilitaron la incorporación de otros agentes antineoplásicos, de carácter más selectivo, como es el caso del bortezomib, un inhibidor del proteasoma 26S, el cual resulta ser una herramienta celular fundamental en el control del ciclo vital celular y de la apoptosis, particularmente en el ámbito tumoral. Bortezomib fue autorizado en 2004 para el tratamiento de los pacientes con MM que han recibido previamente al menos dos tratamientos y que presentan progresión de la enfermedad demostrada con el último de estos tratamientos. Los datos clínicos disponibles indican unas tasas de respuesta del orden del 35% (incluyendo respuestas completas y parciales), con tiempos de progresión de la enfermedad de 9 a 13 meses para los pacientes respondedores, bastante superiores a los previamente descritos en la bibliografía para pacientes con MM refractario o recidivante (alrededor de 3 meses).
El carfilzomib supone una evolución en el ámbito de los inhibidores del proteasoma. Su efecto irreversible y su mayor selectividad sobre la función enzimática de tipo quimotripsina del proteasoma en relación a su antecesor (bortezomib), se tradujo en un efecto antitumoral más potente y eficaz (con mayor supervivencia libre de progresión y reducción de la tasa de progresión tumoral) que el de este, pero también algo más tóxico.
Por otra parte, algunos estudios mostraron que la talidomida es capaz de producir una significativa respuesta en pacientes con mieloma, con tasas de respuesta del orden del 25% en casos recidivantes/refractarios, en su mayoría después de un doble trasplante. A pesar de ser autorizado como medicamento huérfano para el MM –en 2006– y seguir disponible actualmente, su toxicidad1 siempre ha sido un factor limitante de su potencial farmacológico. De ahí, la necesidad de fármacos que mantuvieran sus propiedades o incluso las ampliaran, pero limitando su perfil toxicológico. En esa línea se desarrollaron derivados inmunomoduladores como la lenalidomida, que no solo mantenían las propiedades de la talidomida sobre diversos biomarcadores implicados en diversas patologías, sino que incluso las ampliaban, mejorando los resultados clínicos en MM, con un perfil toxicológico algo más benigno.
La pomalidomida es un análogo de talidomida y lenalidomida (de hecho, se trata del oxoderivado de la lenalidomida), que se emplea en pacientes adultos con MM resistente al tratamiento o recidivante que hayan recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que hayan experimentado una progresión de la enfermedad en el último tratamiento. Presenta una actividad antimielomatosa mayor que sus antecesores, con un perfil toxicológico similar o incluso algo más favorable.
Un aspecto confuso del tratamiento de esta enfermedad ha sido el descubrimiento de que una disminución de los niveles de la proteína mielomatosa en el suero y/o en la orina no se traslada necesariamente a una remisión o aumentos de la supervivencia. Dado que ningún tratamiento actual erradica todas las células mielomatosas, las características de aquellas residuales tras la quimioterapia inicial son de particular importancia. En este sentido, unas pocas células agresivas residuales pueden causar más problemas que una gran cantidad de células inactivas.
Una de las vías farmacológicas que despertó mayor interés en el campo de la terapéutica del MM es la de la molécula CD38, una glicoproteína transmembrana intensamente expresada por células del mieloma y, en menor medida, por diversos tipos de células linfoides y mieloides, e incluso en tejido no hematopoyéticos. A partir de esas observaciones surgió daratumumab, un anticuerpo monoclonal humano que se une específicamente a CD38 e impide sus acciones biológicas, activando diversos mecanismos citotóxicos e inmunitarios que acaban con la lisis o la apoptosis de las células que lo expresan, probablemente a través de citotoxicidad dependiente del complemento, citotoxicidad mediada por anticuerpos y fagocitosis celular dependiente de anticuerpos. Reduce el número de células cancerígenas, pero también se ven afectadas células T y B reguladoras normales y las células citotóxicas NK2, aunque ello no impide que el recuento de linfocitos T en sangre periférica y médula ósea aumente significativamente, lo cual sugiere efectos inmunomoduladores que podrían contribuir a la respuesta clínica. Fue autorizado en 2016 y, desde entonces, ha exhibido eficacia significativa en diversas situaciones clínicas y se han ido ampliando sus indicaciones autorizadas en MM, en combinación con otros fármacos: a) con lenalidomida y dexametasona o con bortezomib, melfalán y prednisona para tratar pacientes adultos de nuevo diagnóstico que no son candidatos a un trasplante autólogo de progenitores hematopoyéticos; b) con bortezomib, talidomida y dexametasona para el tratamiento de pacientes adultos de nuevo diagnóstico candidatos a trasplante; c) con lenalidomida y dexametasona, o bortezomib y dexametasona, para el tratamiento de pacientes adultos pre-tratados con ≥ 1 tratamiento previo. Incluso se ha autorizado en monoterapia para el tratamiento de pacientes adultos con MM en recaída y refractario al tratamiento, que hayan recibido previamente un inhibidor del proteasoma y un agente inmunomodulador y cuya enfermedad haya progresado.
Por otro lado, elotuzumab es un anticuerpo monoclonal dirigido específicamente al SLAMF73 (CS1) que presenta un doble mecanismo de acción: por un lado, activa directamente a las células NK a través tanto de SLAMF7 como de los receptores Fc, potenciando de esta manera la actividad anti-mieloma y, por otro, al unirse al SLAMF7 de la célula mielomatosa, facilita su interacción con las células NK para mediar su destrucción, fundamentalmente a través de la citotoxicidad celular dependiente de anticuerpos. Ha sido el último principio activo comercializado en España con indicación en MM, concretamente, en combinación con lenalidomida y dexametasona para el tratamiento de adultos que han recibido ≥ 1 un tratamiento previo. En comparación con el uso del doblete de lenalidomida+dexametasona solo, la combinación con elotuzumab ha demostrado que reduce un 32% el riesgo de progresión y aumenta la mediana de SLP y SG en más de 4 meses (18,5 vs. 14,3 meses de SLP y 43,7 vs. 39,6 meses de SG), aumentando también la tasa de respuesta global (79% vs. 66%).
En paralelo a estos dos primeros fármacos de inmunoterapia, se aprobó en Europa panobinostat, aún no comercializado en España. Se trata de un inhibidor potente de histona deacetilasa (HDAC) activo por vía oral. La HDAC es una enzima que cataliza la eliminación de los grupos acetilo de los residuos de lisina de histonas y de algunas otras proteínas, por lo que su inhibición provoca una mayor acetilación de las histonas: una alteración epigenética que resulta en una relajación de la cromatina, y que activa la transcripción. Se ha descrito que este fármaco provoca la acumulación de histonas acetiladas y de otras proteínas, y promueve la expresión incrementada del gen supresor del tumor p21CDKNIA (inhibidor de la cinasa dependiente de ciclina 1/p21), mediador clave en el arresto de G1 y en la diferenciación; así, produce la detención del ciclo celular y/o apoptosis de algunas células transformadas, con una mayor citotoxicidad en células tumorales que en células normales. Panobinostat está indicado, en combinación con bortezomib y dexametasona, para el tratamiento de adultos con MM refractario y/o en recaída pre-tratados con ≥ 2 líneas previas con bortezomib y un agente inmunomodulador. Demostró una eficacia notable en un ensayo clínico que comparó la combinación autorizada frente a bortezomib+dexametasona: la triple terapia redujo a la mitad el riesgo de progresión del MM, con un incremento significativo de la mediana de SLP (12,5 meses vs. 4,7 meses) y de la tasa de respuesta global (59% vs. 39%).
A la vista de todas las opciones terapéuticas, se acepta que debe iniciarse el tratamiento en pacientes con MM activo que cumplan los siguientes criterios: hipercalcemia (> 11,0 mg/dl), insuficiencia renal (aclaramiento de creatinina > 2 mg/ml), anemia (Hb < 10 g/dl) y lesiones óseas activas; también si presentan hiperviscosidad sintomática, infecciones bacterianas recurrentes y amiloidosis con afectación orgánica.
Las opciones de tratamiento de primera línea (Figura 1) incluyen las terapias más nuevas: inhibidores de proteasoma y/o fármacos inmunomoduladores, seguido –si está indicado– de un trasplante autólogo de células progenitoras hematopoyéticas (TASPE). La asociación de altas dosis de quimioterapia (AD) con el TASPE ha demostrado mejorar tanto las tasas de respuesta como las expectativas de vida en los pacientes con MM. Se han alcanzado tasas de remisión completa que oscilan entre un 25% y un 75%. La radioterapia juega también un papel importante en el tratamiento inicial del MM, pudiendo ser especialmente efectiva en aquellos pacientes con importante destrucción ósea, intenso dolor y/o compresión nerviosa o de la médula espinal; su mayor desventaja es que daña en forma permanente las células progenitoras hematopoyéticas normales de la médula ósea en el área tratada.
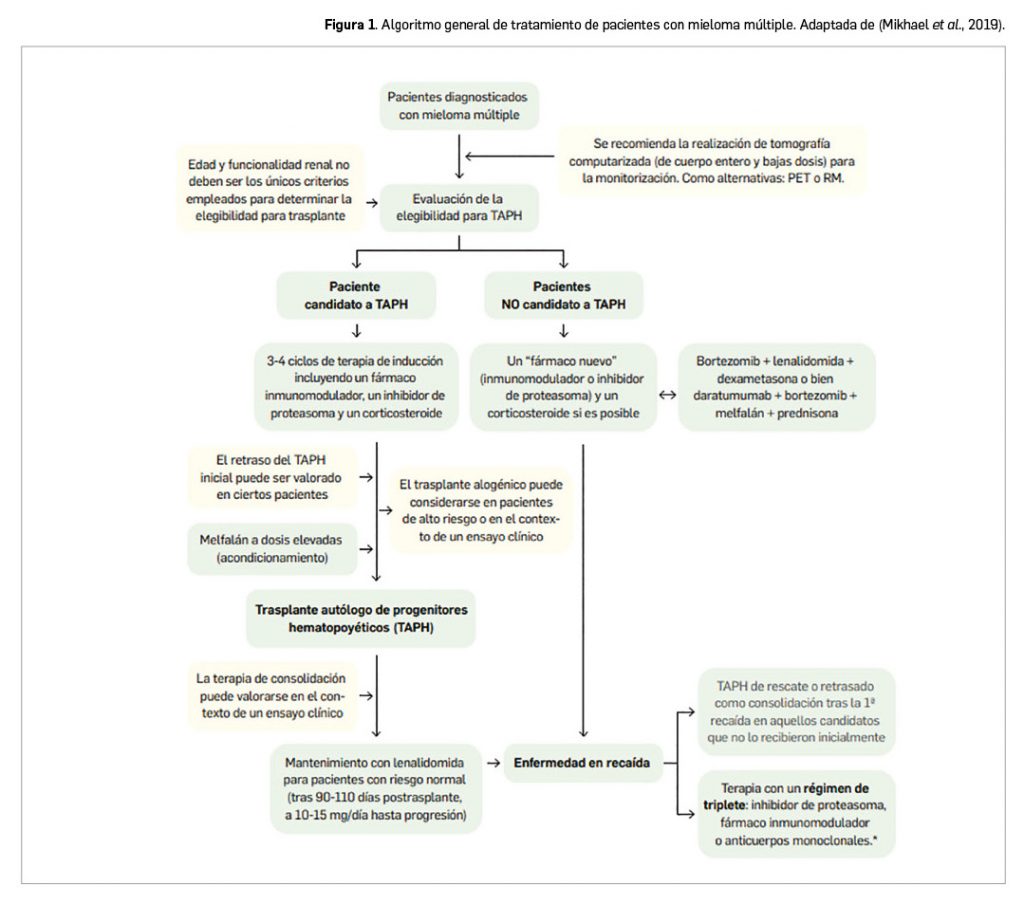
La profundidad de la respuesta al tratamiento de inicio después del trasplante autólogo parece estar correlacionada con la duración del control de la enfermedad hasta la progresión y con la necesidad de una terapia de rescate. En todo caso, el 90% de los pacientes recaen tras un primer tratamiento, con tiempos medios de recaída de entre 1 y 3 años, y la expectativa global de vida es de aproximadamente 4 a 5 años. Las recaídas son, pues, un problema muy frecuente y sobrevienen a pesar de que el interferón alfa o la prednisona en el tratamiento de mantenimiento han probado cierta utilidad para prolongar el periodo de remisión inicial.
Ante una recaída del MM, se requiere una reinducción4. Los regímenes basados en bortezomib y lenalidomida son los más comúnmente usados en combinación con corticosteroides, a los que a veces también se les agrega un agente alquilante o una antraciclina. Con la introducción de las nuevas terapias en las últimas décadas (por ejemplo, carfilzomib, elotuzumab o daratumumab), la mediana de supervivencia global ha mejorado hasta 60 meses desde el diagnóstico de la enfermedad. A pesar de la mejora también en supervivencia libre de progresión en casos de MM recidivante temprano, el 40-60 % de los pacientes no responden a la terapia y casi todos recaen después de uno de estos regímenes (Fernández-Moriano, 2020).
Se acepta que el mieloma reaparece con mayor agresividad tras una recaída, y las reinducciones de respuesta suelen durar menos tras cada recaída, lo que limita la supervivencia. A diferencia de la primera línea, no existe un algoritmo simple para la elección perfecta de la terapia de 2ª, 3ª y 4ª líneas, sino que se deben considerar múltiples factores relacionados con el paciente (estado de salud, comorbilidades, tolerancia y respuesta a tratamientos previos), la enfermedad (tiempo desde la última recaída) y las terapias previas al decidir el tratamiento del MM en recaída.
En ese contexto, es preciso destacar la reciente autorización (2022) de la primera terapia CAR-T autorizada en la UE: idecabtagén vicleucel. Se trata de un tratamiento a base de linfocitos T autólogos modificados para expresar un receptor de antígeno quimérico dirigido específicamente frente al antígeno de maduración de linfocitos B (BCMA), el cual se expresa en la superficie de las células plasmáticas normales y malignas. Está indicado para el tratamiento de pacientes adultos con MM en recaída y refractario que han recibido al menos 3 tratamientos previos, incluidos un agente inmunomodulador, un inhibidor del proteosoma y un anticuerpo anti-CD38 y han presentado progresión de la enfermedad tras el último tratamiento. Ha mostrado resultados muy prometedores que pueden hacer cambiar el enfoque terapéutico en pacientes muy refractarios: un estudio principal con 140 pacientes ha evidenciado tasas de respuesta global del 67%, de las cuales son respuestas completas hasta en el 30% de los pacientes.
Acción y mecanismo
Isatuximab es un nuevo anticuerpo monoclonal que se une específicamente a un epítopo extracelular del receptor CD38, que es expresado de forma altamente uniforme e intensa por las células del mieloma múltiple. Así, activa diversos mecanismos citotóxicos e inmunitarios que acaban con la lisis o apoptosis de dichas células y permiten controlar el crecimiento tumoral. El medicamento ha sido autorizado, en combinación con pomalidomida y dexametasona, para el tratamiento de pacientes adultos con MM resistente al tratamiento o recidivante que han recibido al menos dos tratamientos previos, incluyendo lenalidomida y un inhibidor del proteasoma y han demostrado progresión de la enfermedad en el último tratamiento; de igual modo, se ha aprobado, en combinación con carfilzomib y dexametasona, para el tratamiento de pacientes adultos con MM que han recibido al menos un tratamiento previo.
La CD38 es una glicoproteína transmembrana que se expresa intensamente en las células del mieloma y, en menor medida, en diversos tipos de células linfoides y mieloides, e incluso en tejido no hematopoyéticos. Ejerce múltiples funciones, incluyendo actividad enzimática, regulación de la adhesión celular mediada por receptores y transducción de señales bioquímicas. La actividad enzimática implica la conversión de factores coenzimáticos, como NAD y NADP en diversos sustratos requeridos para la regulación de la señalización intracelular del calcio, como la ADP-ribosa cíclica (se encarga de movilizar iones Ca2+). Además, interviene en la adhesión al endotelio vascular, donde se acopla a la CD31 –su ligando natural presente en la superficie de las células endoteliales– y juega un papel relevante en la migración de los linfocitos. La interacción CD38/CD31 parece ser determinante en la señalización transmembrana, caracterizada por la movilización del Ca2+ y la secreción de diversas citocinas; en concreto, la unión de la CD38 da lugar a la activación de linfocitos T, que induce la secreción de IL-6 e IL-10, factores estimulantes de colonias de granulocitos y macrófagos (GM-CSF) e interferón gamma (IFN-γ).
Se entiende, por tanto, que al inhibir las citadas funciones de CD38, isatuximab tiene un importante papel sobre el crecimiento tumoral. En estudios in vitro se ha probado que ejerce su actividad antineoplásica a través de mecanismos dependientes de las regiones Fc de la inmunoglobulina (IgG), incluyendo la citotoxicidad celular dependiente de anticuerpos (CCDA), la fagocitosis celular dependiente de anticuerpos (FCDA) y la citotoxicidad dependiente del complemento5 (CDC). Además, isatuximab también puede desencadenar la muerte de las células tumorales induciendo la apoptosis a través de un mecanismo independiente de Fc. En ausencia de células tumorales diana que sobre-expresen CD38, el fármaco puede activar las células NK, que expresan los mayores niveles de dicho receptor.
De igual modo, se ha demostrado in vitro que la combinación de isatuximab y pomalidomida induce una mayor lisis de las células mielomatosas CD38+ (por las células efectoras a través de CCDA y la eliminación directa de las células tumorales) respecto al efecto que tiene la monoterapia con isatuximab. Adicionalmente, los estudios in vivo con modelos murinos han probado que ese tratamiento combinado induce una mayor actividad antitumoral sobre xenoinjertos humanos que el uso aislado de cualquiera de los dos fármacos (AEMPS, 2020).
Por último, los estudios clínicos han puesto de manifiesto que en pacientes tratados con el fármaco se aprecia una reducción de las cifras absolutas de células NK totales CD16+ y CD56+, de linfocitos B CD19+, de linfocitos T CD4+ y de linfocitos Th en sangre periférica. Ese tratamiento induce igualmente la expansión clonal del repertorio de receptores de linfocitos T, lo cual es indicativo de una respuesta inmunitaria adaptativa.
Aspectos moleculares
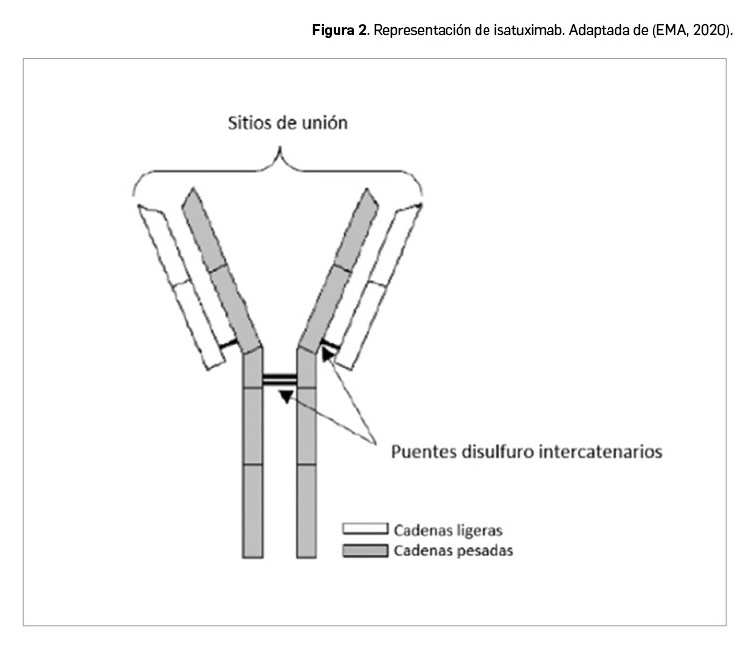
El nuevo fármaco es una inmunoglobulina quimérica de tipo IgG1 derivada de un anticuerpo monoclonal, que se obtiene por ingeniería genética a partir de células derivadas de ovario de hámster chino (línea celular CHO).
La proteína terapéutica está compuesta por dos cadenas ligeras de tipo kappa, cada una de ellas compuesta por 214 residuos aminoacídicos y con un peso molecular de aproximadamente 23 kDa, y dos cadenas pesadas de tipo IgG1, cada una formada por 450 aminoácidos que totalizan 49 kDa en su forma no glicosilada; las cadenas pesadas se unen entre sí por puentes disulfuro (Figura 2). Entre las especificidades moleculares, se puede destacar que el residuo de glutamina en el extremo N-terminal de las cadenas pesadas se convierte completamente en piroglutamato, y la mayoría de los extremos C-terminales de las cadenas pesadas se recortan. Isatuximab contiene 32 residuos de glutamina N-terminal (que establecen 16 puentes disulfuro) y dos sitios de glicosilación localizados en los residuos de asparagina en posición 300 de cada cadena pesada.
Eficacia y seguridad clínicas
La eficacia y la seguridad clínicas de isatuximab por vía intravenosa han sido adecuadamente contrastadas en las indicaciones y dosis autorizadas mediante dos ensayos clínicos pivotales confirmatorios de fase 3 de similar diseño –aleatorizados, multicéntricos y multinacionales, abiertos y de 2 brazos de tratamiento– en pacientes adultos con mieloma múltiple resistente al tratamiento y/o recidivante.
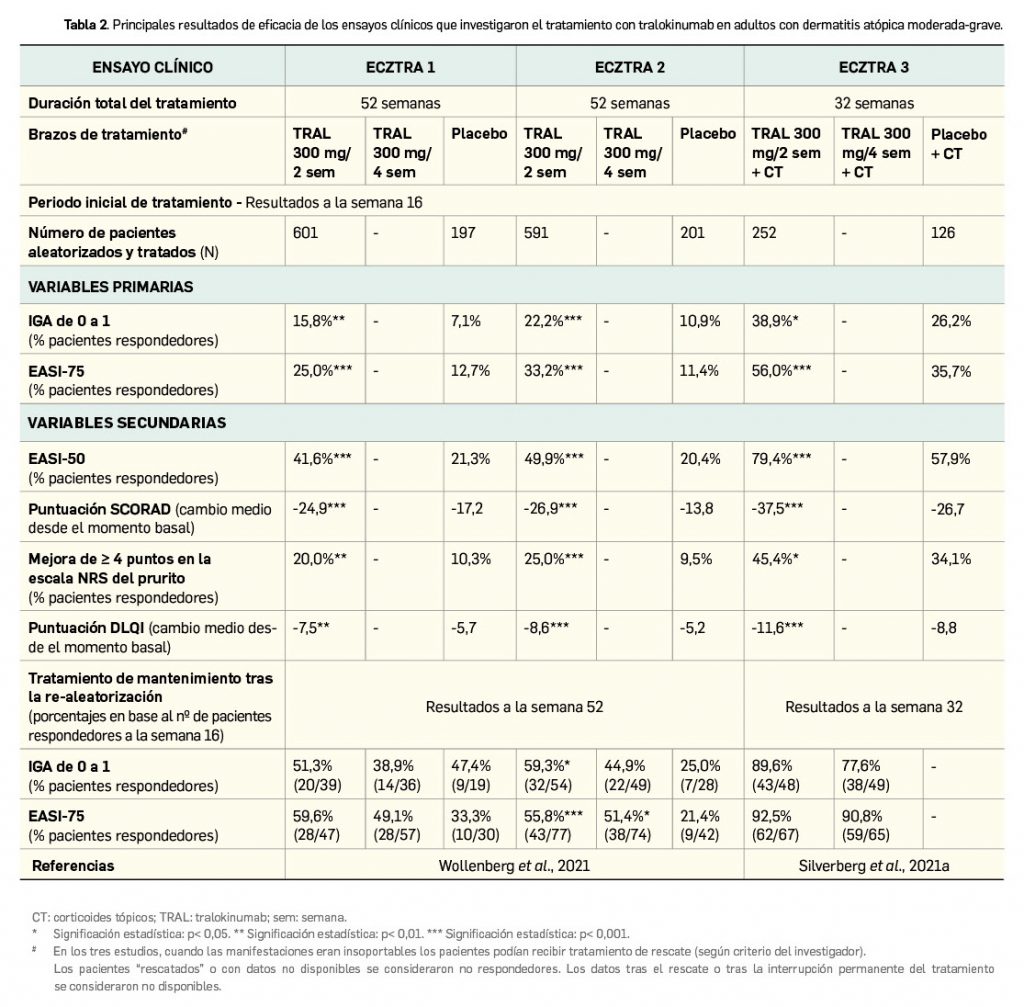
Su primera indicación (uso combinado con pomalidomida y dexametasona en tercera línea de tratamiento o posterior) se sustentó en el estudio ICARIA-MM (Richardson et al., 2021), desarrollado en 111 centros de 24 países, que aleatorizó (1:1) a un total de 307 pacientes a recibir la combinación de isatuximab (volumen basado en el peso) con pomalidomida oral y dexametasona oral/intravenosa (IPD; n= 154) o bien solo pomalidomida y dexametasona (PD; n= 153) como control activo en ciclos de 28 días, hasta progresión de la enfermedad o toxicidad inaceptable. Los pacientes debían haber recibido un mínimo de 2 tratamientos previos, incluyendo lenalidomida y un inhibidor de proteasoma, y su MM debía estar en progresión en los 2 meses siguientes.
A grandes rasgos, las características demográficas y clínicas basales de los participantes estuvieron bien equilibradas en los dos grupos. Entre ellas destacan las siguientes: la mediana de edad de los pacientes fue de 67 años (el 20% tenía ≥ 75 años), el estado funcional de los pacientes era mayoritariamente bueno (ECOG 0 en el 36% de los pacientes del grupo de isatuximab y en el 45% de los del grupo control, y ECOG 1 en el 54% y 44%, respectivamente), casi un 20% presentaban anomalías cromosómicas de alto riesgo (sobre todo, del 17p y t 4;14) y aproximadamente un tercio de los pacientes en ambos grupos tenía insuficiencia renal. El mieloma estaba en estadio I en el 38% de los pacientes, II en el 36% y III en el 25%; la mediana de líneas de tratamiento previo fue de 3 (todos los pacientes habían sido pretratados con un inhibidor de proteasoma y lenalidomida, y eran mayoritariamente resistentes a ellos), y un 56% había recibido un trasplante de células madre.
Con una mediana de duración del tratamiento de 41 semanas en el brazo experimental y de 24 semanas en el brazo comparador, la supervivencia libre de progresión (SLP) evaluada por un comité independiente –variable principal– se prolongó significativamente con la adición de isatuximab, alcanzando una mediana de 11,53 meses (IC95% 8,9-13,9) en su grupo frente a los 6,47 meses (IC95% 4,5-8,3) en el grupo control (HR= 0,59; IC95% 0,44-0,81; p= 0,001). La tasa de respuesta global también creció notablemente hasta el 60,4% (93/154), respecto al 35,3% (54/153) con el comparador activo (OR= 2,8; IC95% 1,7-4,6; p< 0,0001); si bien las respuestas completas fueron escasas en ambos grupos (4,5% vs. 2%) y las respuestas parciales se mantuvieron en niveles similares (28,6% vs. 26,8%), la tasa de muy buena respuesta parcial fue superior en el brazo de isatuximab (27,3% vs. 6,5%). La duración de la respuesta fue de 13,3 meses en ese grupo y de 11,1 meses en el grupo control, siendo el inicio de la respuesta más rápido –casi la mitad– en los pacientes respondedores del brazo experimental (mediana de tiempo hasta respuesta de 35 días vs. 58 días en el grupo control). Los análisis por subgrupos de pacientes respaldan la eficacia consistente de isatuximab con independencia de los factores pronóstico (Richardson et al., 2022). Así, por ejemplo, se verificaron mejoras de la SLP con la adición de isatuximab a pomalidomida+dexametasona en los pacientes con características citogenéticas de alto riesgo (mediana de 7,5 meses vs. 3,7 meses en el grupo control; HR= 0,66; IC95% 0,3-1,3), en pacientes de ≥ 75 años de edad (HR= 0,48; IC95% 0,2-,9), con enfermedad basal en estadio III (HR= 0,64; IC95% 0,4-1,1), con insuficiencia renal (HR= 0,50; IC95% 0,3-0,8), con > 3 líneas previas de tratamiento (HR= 0,59; IC95% 0,4-0,98), y en pacientes resistentes a lenalidomida (HR= 0,59; IC95% 0,4-0,8) o a un inhibidor del proteasoma (HR= 0,58; IC95% 0,4-0,8).
Con una mediana de seguimiento de los pacientes de 35 meses, el análisis provisional de la supervivencia global (SG) revela que alcanzó una mediana de 24,6 meses (IC95% 20,3-31,3) en el grupo de isatuximab, superior a los 17,7 meses del grupo control (IC95% 14,4-26,2), si bien la diferencia no tuvo significación estadística (HR= 0,76; IC95% 0,6-1,0; p= 0,028). El análisis final sobre SG está aún en marcha.
Por otra parte, la segunda indicación de isatuximab en MM (en segunda línea de tratamiento o posterior asociado a carfilzomib y dexametasona) se basó en los resultados del estudio IKEMA, realizado en 70 centros de 16 países, que aleatorizó (3:2) a 302 pacientes a recibir isatuximab en combinación con dexametasona y carfilzomib (n= 179) o bien solo carfilzomib con dexametasona como control (n= 123) en ciclos de 28 días hasta progresión de la enfermedad o toxicidad inaceptable. Los pacientes debían haber recibido entre 1 y 3 terapias previas, si bien se excluyeron aquellos con enfermedad primaria refractaria o pretratados con carfilzomib o daratumumab.
De nuevo, las características demográficas y clínicas de los pacientes eran similares en ambos grupos. La mediana de edad de los pacientes fue de 64 años (9% con ≥ 75 años), una amplia mayoría de ellos tenía un buen estado funcional (ECOG 0-1 en el 94% en el grupo de isatuximab y 96% en el grupo control) y el estadio del MM en el momento basal era I en el 53%, II en el 31% y III en el 15%; un cuarto de los pacientes presentaba anomalías cromosómicas de alto riesgo (sobre todo, del 17p y t 4;14) y aproximadamente un 20% sufría insuficiencia renal. En cuanto a los tratamientos previos, la mediana de líneas terapéuticas fue de 2, con un 44% de los pacientes que solo había recibido 1 línea; casi todos habían recibido inhibidores del proteasoma (90%; 33% eran refractarios) e inmunomoduladores (78%; 45% eran refractarios), y el 61% había sido sometido a trasplante de células madre.
Con una duración media del tratamiento de 80 semanas en el brazo experimental y de 61 semanas en el del comparador, la SLP –variable primaria, evaluada por comité independiente– se prolongó significativamente con isatuximab: no se alcanzó la mediana en el momento de corte de datos frente a una mediana de 19,15 meses en el grupo control (HR= 0,53; IC95 0,32-0,89; p= 0,0013). Si bien la tasa de respuesta global fue pareja en ambos grupos (86,6% vs. 82,9%; p= 0,39), la frecuencia de muy buena respuesta parcial o mejor fue superior en el brazo experimental (72,6% vs. 56,1%; p= 0,0021), incluyendo la tasa de respuesta completa (39,7% vs. 27,6%); ello se correlacionó con una mayor tasa negativa de enfermedad residual mínima (29,6% vs. 13,0%; 0,0008). La mediana de duración de la respuesta no se alcanzó en ninguno de los dos grupos y el tiempo hasta la respuesta en los pacientes respondedores fue similar con ambos tratamientos: en torno a 1,1 mes. No se tienen datos maduros de SG, pero parece que la tendencia puede ser favorable a la combinación con el nuevo fármaco: con una mediana de seguimiento de 21 meses, la tasa de mortalidad era del 17%, frente al 20% en el grupo control.
Los análisis por subgrupos también respaldan la eficacia consistente de isatuximab en esta indicación. Así, se confirmó un beneficio en SLP en pacientes con citogenética de alto riesgo (HR= 0,72), con anomalías cromosómicas (HR= 0,57), con edad de ≥ 65 años (HR= 0,43), con insuficiencia renal (HR= 0,27), con más de 1 línea de tratamiento previo (HR= 0,48), con estadio III al inicio (HR= 0,65) y en pacientes refractarios a lenalidomida (HR= 0,60).
Con respecto a la seguridad clínica, el perfil toxicológico de isatuximab ha sido adecuadamente definido en su desarrollo clínico y, aunque importante, parece clínicamente manejable. La práctica totalidad de los pacientes notifica algún evento adverso durante el tratamiento, graves en una alta proporción (en torno al 60%), lo que lleva a una tasa no desdeñable de discontinuaciones por motivos de seguridad (7%).
En su combinación con pomalidomida y dexametasona (duración media de exposición de 41 semanas), las reacciones adversas más frecuentes en comparación con el control fueron: neutropenia (47%), reacciones a la perfusión (38%), neumonía (31%), infección del tracto respiratorio superior (28%), diarrea (26%) y bronquitis (24%); entre las reacciones adversas graves (62%) sobresalen neumonía (26%) y neutropenia febril (7%). En su asociación a carfilzomib y dexametasona (duración media de exposición de 80 semanas), la afectación de los recuentos celulares fue menor, pero el resto de las reacciones adversas más frecuentes fueron parecidas: reacciones a la perfusión (46%), hipertensión (37%), diarrea (36%), infección del tracto respiratorio superior (36%), neumonía (29%), fatiga (28%), disnea (28%), insomnio (24%), bronquitis (23%) y dolor de espalda (22%); entre las de mayor severidad (59% de grado ≥ 3) también sobresale la neumonía (22%). Los citados eventos adversos tuvieron desenlace fatal en el 8% de los pacientes que recibieron isatuximab+pomalidomida+dexametasona y en el 3% de los tratados con isatuximab+carfilzomib+dexametasona, destacando como causas la neumonía y otras infecciones graves. No parece que la baja tasa de inmunogenicidad mostrada por el fármaco en 9 estudios con pacientes con MM (2% de prevalencia de anticuerpos anti-fármaco) tenga relevancia clínica (AEMPS, 2020).
Aspectos innovadores
Isatuximab es un nuevo anticuerpo monoclonal de tipo IgG1 que se une específicamente al receptor CD38, el cual se expresa uniforme e intensamente por las células del mieloma múltiple (MM). Esa unión media la activación de diversos mecanismos citotóxicos e inmunitarios –dependientes de la región Fc del anticuerpo (como la citotoxicidad celular o la fagocitosis dependientes de anticuerpos o la citotoxicidad dependiente del complemento) o independientes de la misma– que acaban con la lisis o apoptosis de las células mielomatosas y permiten controlar el crecimiento tumoral. El medicamento ha sido autorizado, en combinación con pomalidomida y dexametasona, para el tratamiento de pacientes adultos con MM resistente al tratamiento o recidivante que han recibido al menos dos tratamientos previos, incluyendo lenalidomida y un inhibidor del proteasoma y han mostrado progresión de la enfermedad tras el último tratamiento; y en combinación con carfilzomib y dexametasona para el tratamiento de pacientes adultos con MM que han recibido al menos un tratamiento previo.
Su aprobación se ha sustentado en dos ensayos clínicos de diseño adecuado, que han permitido confirmar su favorable balance beneficio-riesgo en una población de pacientes representativa de la población diana del medicamento. El primero de ellos (ICARIA-MM) demostró la superioridad de la adición de isatuximab a la combinación de pomalidomida y dexametasona, en comparación con el solo uso de estos dos fármacos como control activo, en pacientes con MM en progresión tras haber recibido al menos 2 líneas terapéuticas previas, incluyendo lenalidomida y un inhibidor de proteasoma. El tratamiento triple durante una mediana de 41 semanas prolongó la mediana de SLP en unos 5 meses (11,5 vs. 6,5 meses), reduciendo el riesgo de progresión de la enfermedad o muerte en un 40%. También mejoró la tasa de respuesta antitumoral, casi duplicándola (60% vs. 35%), así como la profundidad de dicha respuesta (muy buena respuesta parcial o mejor en el 32% vs. 9%). Todo ello resultó en una tendencia favorable en SG –prolongación de la mediana en casi 7 meses (24,6 vs. 17,7)– que aún debe confirmarse con los análisis finales.
El segundo estudio (IKEMA) probó que la asociación del nuevo fármaco a la combinación de carfilzomib y dexametasona es también superior al solo uso de estos dos últimos en pacientes con MM en recaída que habían recibido entre 1 y 3 líneas previas de tratamiento (excluyendo carfilzomib y daratumumab). La administración de la triple terapia durante una mediana de 80 semanas supuso una prolongación notable de la SLP (no se alcanzó la mediana en el brazo experimental vs. 19,1 meses en el control) y una reducción del 47% del riesgo de progresión o muerte. Con tasas de respuesta parecidas en ambos grupos, la profundidad de la misma fue significativamente mayor en los pacientes tratados con isatuximab: muy buena respuesta parcial o mejor en el 73% vs. 56%. Tras una mediana de 21 meses de seguimiento, no se tienen datos maduros de SG, de modo que no se puede concluir al respecto.
En ambos estudios se verificó que las combinaciones que incorporan el nuevo fármaco ejercen una eficacia rápida (respuesta objetiva en aproximadamente 1 mes desde el inicio de tratamiento), duradera y consistente, con independencia de factores como la edad, el riesgo citogenético, la funcionalidad renal, el estadio del MM, las líneas de tratamiento previos o la refractariedad a fármacos concretos.
Por otra parte, se ha caracterizado adecuadamente su perfil toxicológico, que es importante y se asocia a una alta frecuencia de reacciones adversas, graves en casi 2 de cada 3 pacientes (≈60%), que suponen una tasa de discontinuaciones reseñable (≈7%). Los eventos adversos de mayor incidencia, en su mayoría manejables en clínica, serían esperables por tratarse de una proteína inmunomoduladora y a la vista de la seguridad de los fármacos con que se combina. Con una escasa inmunogenidad, sobresalen por su frecuencia (≥ 20%) las infecciones del tracto respiratorio superior e inferior (incluidas bronquitis y neumonía), citopenias (sobre todo, neutropenia), reacciones a la perfusión, alteraciones del tracto gastrointestinal (náuseas y diarrea) y otras inespecíficas, como fatiga o insomnio. Por su gravedad, las infecciones y, en particular, la neumonía, parecen constituir el mayor riesgo asociado a isatuximab.
Habida cuenta del carácter por ahora incurable del mieloma y de las frecuentes recaídas de los pacientes, casi todos van pasando por sucesivas líneas de tratamiento6. Se acepta que el uso de 2 o 3 fármacos juntos, incluyendo casi siempre un inmunomodulador o inhibidor de proteasoma (bortezomib y/o lenalidomida), aporta mejores resultados clínicos en comparación con las monoterapias como alternativas de rescate en casos en recaída/refractariedad. Las principales guías clínicas apuntan a la falta de respuesta a lenalidomida como el aspecto más relevante que orienta hacia una pauta u otra.
Isatuximab no supone ninguna novedad en cuanto a mecanismo de acción, sino que comparte vía terapéutica con el ya comercializado daratumumab, pese a que se unen a distintos epítopos de CD38 e inducen distintos cambios conformacionales en esta molécula. No se dispone de comparaciones directas entre ambos fármacos para conocer si esas pequeñas diferencias tienen implicación clínica. Un meta-análisis que revisó datos de 13 estudios de fase 3 en pacientes pretratados sugirió que daratumumab y doxorrubicina liposomal pegilada tienen la mayor probabilidad de alcanzar mejoras en SPL, seguidos de isatuximab, carfilzomib, pomalidomida y panobinostat, sin disponerse de datos maduros de SG; también apunta a que la incidencia de eventos adversos graves es mayor con isatuximab, seguido de panobinostat y pomalidomida (Acuri et al., 2021). Así pues, pese a que se trata de una evidencia de robustez limitada, no parece que isatuximab vaya a superar las mejoras terapéuticas incorporadas previamente por daratumumab, el cual, además, tiene un espectro de indicaciones autorizadas más amplio. Cabe destacar que el empleo de daratumumab en 1ª línea es muy relevante y se considera de elección su inclusión junto a otros agentes en el esquema de rescate: el hecho de que no se disponga de datos suficientes sobre la eficacia de isatuximab en pacientes pretratados con daratumumab limita su uso.
En definitiva, se ha probado sólidamente la superioridad de la asociación de isatuximab a dos combinaciones de fármacos (pomalidomida+dexametasona y carfilzomib+dexametasona) previamente aprobadas como terapias de rescate en pacientes pretratados con MM en recaída. El nuevo fármaco representa, pues, una alternativa más de triple terapia a partir de la 2ª línea de tratamiento, y su uso puede ser especialmente relevante en pacientes refractarios a lenalidomida; en ese contexto, se suma a otros fármacos biológicos ya disponibles (por ejemplo, daratumumab o elotuzumab) sin aportar aparentemente ningún elemento de innovación terapéutica disruptiva (SEHH, 2021). Es preciso recordar que en este contexto la futura disponibilidad de las prometedoras terapias CAR-T (concretamente, el anti-BCMA idecabtagén vicleucel) hace previsible un cambio en los enfoques de tratamiento.
Valoración

Fármacos relaciones registrados en España