Número 452, Abril 2022


Además de la información que se incluye en los listados mensuales publicados en PAM, en BOT PLUS se incluye un apartado de Histórico, en las fichas de medicamentos, en el que se presenta información referente a cambios que haya sufrido anteriormente el medicamento o producto, entre otros, los cambios de nombre y los cambios de laboratorio. Esta información también está disponible para productos sanitarios financiados o dietoterápicos.
Se añade la posibilidad de visualización de las situaciones anteriores (o incluso futuras) relacionadas con un cambio de nombre. Con automatismos que nos permiten localizar un medicamento que haya cambiado de nombre, independientemente de cuál usemos.

Además de la información existente en Histórico, se permite la explotación de la información incluida en BOT PLUS en este apartado, mediante la integración de la información almacenada en Histórico en el apartado de Listados de BOT PLUS, que permite realizar consultas entre rangos de fechas y por un concepto en concreto de entre los almacenados en el apartado de Histórico. Entre ellos se incluyen, precisamente, los conceptos “Cambio del nombre del medicamento” y “Cambio del laboratorio comercializador”.

Resumen delas notas sobre seguridad y farmacovigilancia publicadas por la AEMPS desde principios del año 2021. Para información más ampliada y acceso al documento de la AEMPS, puede consultar BOT PLUS.

Alertas debidas de calidad observados en medicamentos de uso humano, publicadas por la AEMPS desde el anterior número y que suponen la retirada o inmovilización de ciertos lotes de medicamentos. En BOT PLUS puede encontrar más información detallada, con acceso al documento de la AEMPS.

Además de los listados mensuales que podemos consultar en PAM, en BOT PLUS se incorpora la información que publica la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) relativa a notificaciones sobre seguridad y/o calidad de los medicamentos. Mediante un pictograma específico se pueden visualizar de forma rápida medicamentos afectados por alguna alerta de seguridad o de defectos de calidad, con tan solo entrar en su ficha.
Al acceder a la ficha de un medicamento afectado por una retirada, se visualiza un mensaje con la advertencia “Medicamento con lotes retirados por Alerta AEMPS. Ver más información en “Alertas Calidad”. Además, se incluye una pestaña específica en la que se pueden consultar los lotes concretos que han sido retirados, con sus respectivas fechas de caducidad, así como la descripción del defecto de calidad detectado y las medidas a adoptar. También se cuenta con acceso al documento publicado por la AEMPS.
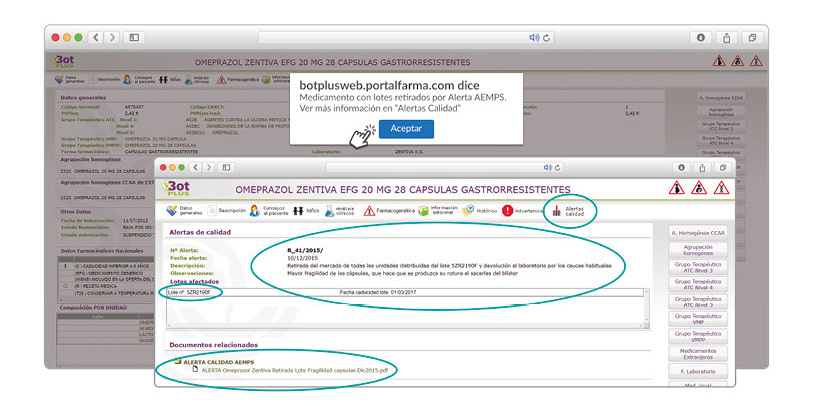
De forma interesante, dicha información se puede explotar a través de la Búsqueda Libre de BOT PLUS para obtener listados de todos los medicamentos afectados por alertas de calidad que implica la retirada (o también la inmovilización) de sus lotes en un momento dado.
Esta codificación de los lotes retirados es una información puesta a disposición de todos los usuarios, con el objetivo de ofrecer una nueva información capaz de integrarse con otros sistemas de información y mejorar la gestión e identificación de estos medicamentos, en los que la labor asistencial y de control del farmacéutico es fundamental.
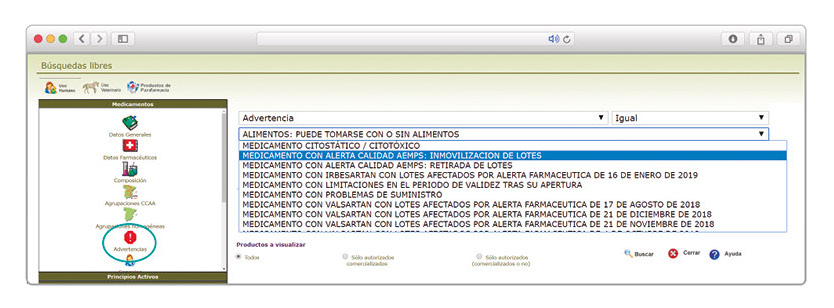
Listado de medicamentos con problemas de suministro publicado por la AEMPS, a fecha de cierre de este número. En BOT PLUS, se puede encontrar la información completamente actualizada, al tratarse de una información que varía de forma continua.


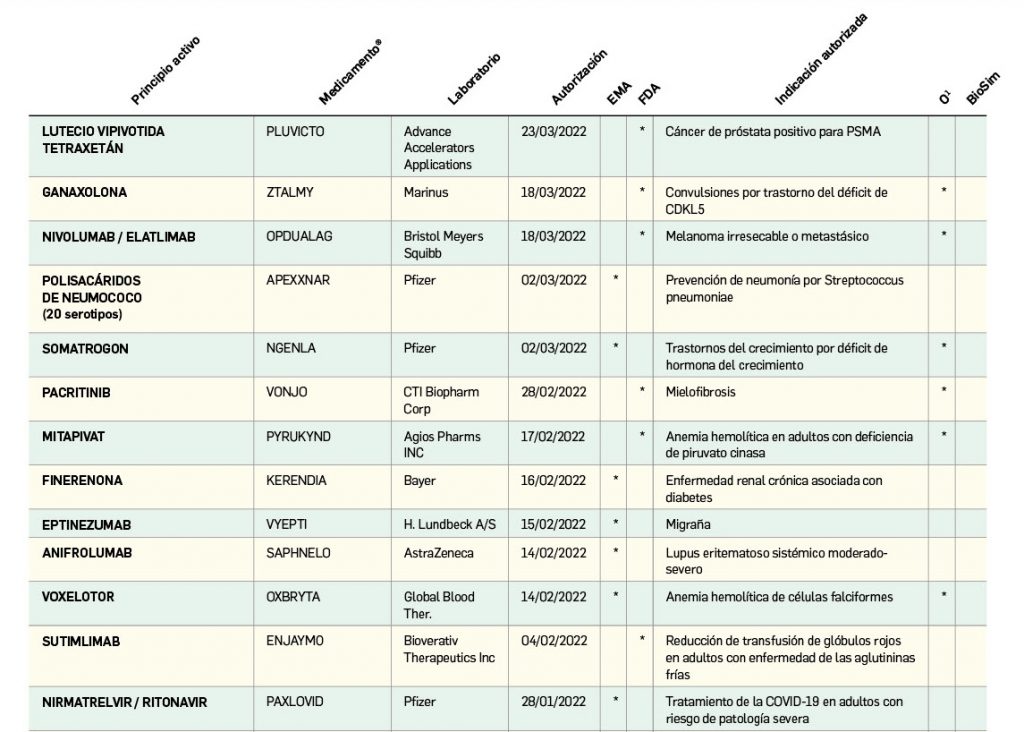
Los medicamentos de terapia avanzada (MTA o Advanced Therapy Medicinal Products, ATMP) ofrecen nuevos e innovadores tratamientos para las enfermedades. Están basados en la terapia génica, la terapia celular somática o la ingeniería tisular. El marco legal para las ATMP en la Unión Europea está establecido en la Regulation (EC) No 1394/2007 on advanced therapy medicinal products que asegura el libre movimiento de estas medicinas dentro de la Unión Europea y el acceso a los mercados. La regulación (EC) nº 1394/2007 también establece el nuevo Comité en Terapias avanzadas (CAT) cuya responsabilidad fundamental consiste en preparar un proyecto de opinión sobre cada nueva solicitud de medicamento de terapia avanzada planteada a la Agencia Europea de Medicamentos, antes de que el Comité de Medicamentos de Uso Humano (CHMP, Committee for Medicinal Products for Human Use) de la misma adopte una opinión definitiva sobre la concesión, modificación, suspensión o revocación de una autorización de comercialización para el medicamento en cuestión.




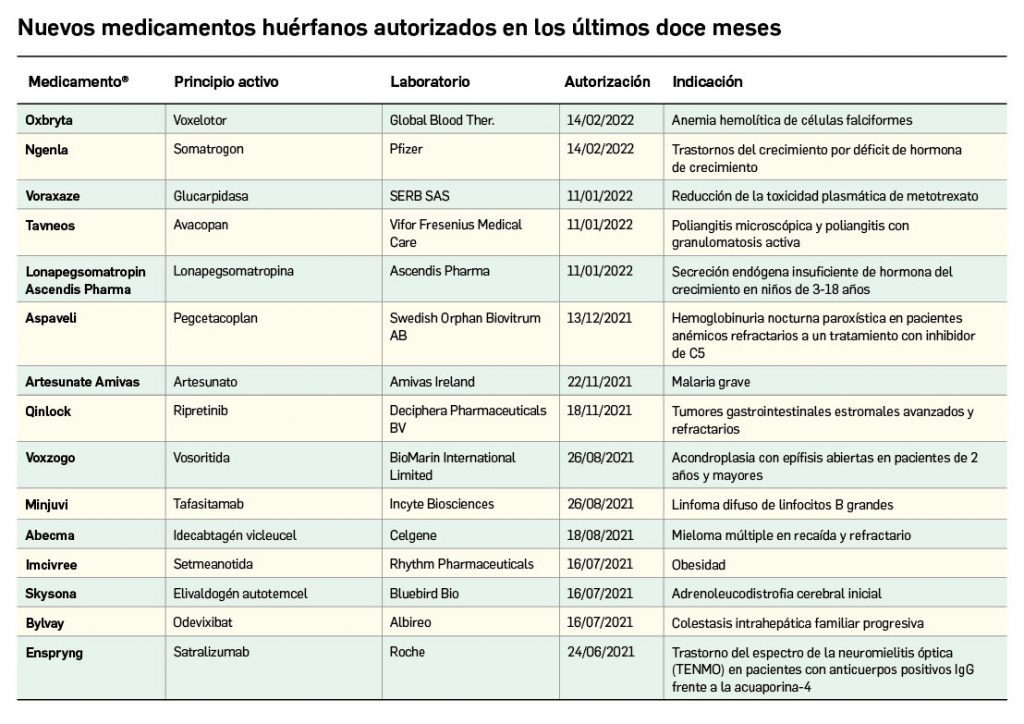
Los medicamentos huérfanos son aquéllos que sirven para diagnosticar, prevenir o tratar enfermedades raras de carácter muy grave o con riesgo para la vida. En la Unión Europea, la calificación de enfermedad rara se aplica a todas aquellas que no afectan a más de 5 de cada 10.000 personas. La designación de un medicamento como huérfano no garantiza su uso en la condición designada y no implica necesariamente que el producto satisfaga los criterios de eficacia, seguridad y calidad necesarios para la concesión de la autorización de comercialización. Como para cualquier medicamento, estos criterios solo pueden ser evaluados una vez que la solicitud de autorización de comercialización haya sido presentada.
► Instituto de Salud Carlos III (Ministerio de Ciencia e Innovación):
Instituto de Investigación en Enfermedades Raras:
https://www.isciii.es/QuienesSomos/CentrosPropios/IIER/Paginas/default.aspx
CIBERER (Centro de Investigación Biomédica en Red de Enfermedades Raras): https://www.ciberer.es/
► Instituto de Mayores y Servicios Sociales (IMSERSO, Ministerio de Derechos Sociales y Agenda 2030):
http://www.imserso.es/imserso_01/index.htm
► Federación Española de Enfermedades Raras (FEDER): www.enfermedades-raras.org
► Asociaciones de pacientes en España: https://enfermedades-raras.org/index.php/asociaciones/nuestros-socios
► Agencia Europea de Medicamentos (EMA; Europea Medicines Agency). Apartado de Medicamentos Huérfanos
(inglés): https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview
https://www.ema.europa.eu/en/committees/committee-orphan-medicinal-products-comp
► Comisión Europea: web oficial de la Comisión Europea sobre enfermedades raras y medicamentos huérfanos (español).
http://ec.europa.eu/health/rare_diseases/policy/index_es.htm
► Orphanet: Portal de información oficial de la Unión Europea sobre enfermedades raras y medicamentos huérfanos (español).
http://www.orpha.net/consor/cgi-bin/index.php?lng=ES
► Eurordis: Federación Europea de Asociaciones de Pacientes con Enfermedades Raras (español). http://www.eurordis.org/es
► Food & Drug Administration (FDA, Estados Unidos). Apartado de Medicamentos Huérfanos (inglés):
http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/default.htm
► Pharmaceuticals & Medical Devices Agency. Agencia de Medicamentos y Dispositivos Médicos, de Japón (inglés):
http://www.pmda.go.jp/english/index.html
La autoridad australiana de medicamentos, Therapeutic Goods Administration (TGA), ha informado de la ampliación del tiempo posterior a un tratamiento con tamoxifeno desde 2 meses a 9 meses, para mantener el uso de métodos anticonceptivos de barrera y así evitar un embarazo.
La autoridad reguladora de medicamentos de Australia, Therapeutic Goods Administration (TGA), ha recomendado ampliar el plazo posterior a un tratamiento con tamoxifeno durante el que se deben evitar embarazos y así el riesgo de malformaciones en el posible feto. Para ello, ha anunciado que la duración de la anticoncepción tras finalizar el tratamiento con tamoxifeno se ha ampliado de 2 meses a 9 meses (TGA, 2021).
El tamoxifeno es un modulador selectivo de los receptores de estrógenos indicado en el tratamiento del cáncer de mama. Inhibe el crecimiento tumoral al competir con el estrógeno por los sitios receptores en el tejido mamario y está también indicado para la reducción primaria del riesgo de cáncer de mama en mujeres con un riesgo moderado o alto.
El fármaco está contraindicado en el embarazo y debe descartarse la posibilidad de embarazo antes de iniciar el tratamiento. Se debe recomendar a las mujeres en edad fértil que utilicen métodos anticonceptivos de barrera u otros métodos anticonceptivos no hormonales si son sexualmente activas, tanto durante el tratamiento como durante los nueve meses posteriores a su finalización.
Se debe informar a las mujeres sobre los riesgos potenciales para el feto en caso de que queden embarazadas mientras toman tamoxifeno o dentro de los nueve meses posteriores a la finalización del tratamiento. La duración recomendada de la anticoncepción después de finalizar el tratamiento con tamoxifeno se ha ampliado de dos a nueve meses. Esto significa que las mujeres deben continuar con su anticoncepción, y no quedar embarazadas, durante al menos nueve meses después de que finalice la terapia con tamoxifeno. No obstante, la categoría australiana de embarazo para el tamoxifeno permanece sin cambios, categoría B3, según la cual se ha registrado un pequeño número de informes de abortos espontáneos, defectos de nacimiento y muertes fetales después de que las mujeres hayan tomado tamoxifeno, aunque no se ha establecido una relación causal.
La decisión se basa en la guía de la FDA de EE.UU. para productos farmacéuticos genotóxicos (FDA, 2019) que recomienda un período mínimo de anticoncepción de seis meses más cinco vidas medias de eliminación, después de la interrupción de la terapia.
En España, la información de las fichas técnicas (FT) de los medicamentos con tamoxifeno (Tamoxifeno Cinfa®, Tamoxifeno Funk®, Tamoxifeno Vir®)
fue actualizada en diciembre de 2021, e indica la siguiente precaución en la sección 4.6. Fertilidad, embarazo y lactancia: “Se deberá advertir a las mujeres de no quedarse embarazadas mientras toman tamoxifeno y deberán usar métodos de barrera u otros métodos anticonceptivos no hormonales si son potencialmente fértiles. Las pacientes premenopáusicas serán examinadas cuidadosamente antes de comenzar el tratamiento, para excluir la posibilidad de embarazo. Igualmente, deberá advertirse a las mujeres de los riesgos potenciales para el feto, si se quedaran embarazadas mientras se les administra tamoxifeno o en un periodo de dos (2) meses desde la suspensión del tratamiento (ver sección 4.4)”.
La duración recomendada en Australia de la anticoncepción de barrera, nunca hormonal, una vez finalizado el tratamiento con tamoxifeno se ha ampliado de dos (2) a nueve (9) meses. Esto significa que las mujeres deben continuar con su anticoncepción de barrera durante todo el tratamiento, y no quedar embarazadas durante al menos nueve meses después de que finalice la terapia con tamoxifeno. Hasta que se modifique de forma efectiva en Europa la precaución actual, se debería seguir la precaución de la agencia australiana, manteniendo la anticoncepción de barrera física, no hormonal, durante 9 meses desde la finalización del tratamiento con tamoxifeno.
La Agencia Española de Medicamentos y Productos Sani- croorganismos vivos (por ejemplo, la vacuna BCG) en los tarios (AEMPS) ha remitido a los profesionales sanitarios, 12 meses después del nacimiento a los lactantes que es- a través de los laboratorios titulares de los medicamentos tuvieron expuestos a infliximab en el útero. De igual modo, con infliximab (Remicade®, Flixabi®, Inflectra®, Remsima®, también se desaconseja la administración de vacunas de Zessly®), la información reciente sobre los niveles plasmámicroorganismos vivos a lactantes alimentados con leche ticos de infliximab en niños nacidos, o lactantes, de madres materna mientras la madre esté recibiendo infliximab, a que reciban tratamiento con infliximab. En base a ella, no ser que los niveles séricos de infliximab en el lactante advierte de que no se deben administrar vacunas de misean indetectables.
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha remitido información a través de los laboratorios titulares de la comercialización de los medicamentos con infliximab (Remicade®, Flixabi®, Inflectra®, Remsima®, Zessly®) mediante una “comunicación directa a profesionales sanitarios” (como DHPC, según siglas del inglés) sobre nuevos datos de seguridad. Todas las 27 agencias reguladoras nacionales europeas han realizado la misma comunicación DHPC, consensuada en el seno del Comité Europeo para la Evaluación de Riesgos en Farmacovigilancia (PRAC), el pasado 7 de marzo de 2022 (AEMPS, 2022; EMA, 2022).
Infliximab (Remicade® y sus biosimilares Flixabi®, Inflectra®, Remsima® y Zessly®) es un anticuerpo monoclonal de tipo IgG1 quimérico murino-humano que se une específicamente al TNFα humano. En la Unión Europea, está indicado para el tratamiento de artritis reumatoide, enfermedad de Crohn (en adultos y en pediatría), espondilitis anquilosante, colitis ulcerosa (en adultos y en pediatría), psoriasis y artritis psoriásica. Respecto a la administración de vacunas basadas en microorganismos vivos a lactantes que estuvieron expuestos a infliximab en el útero, hay que tener en cuenta que el fármaco atraviesa la placenta, y se ha detectado en el suero de lactantes que estuvieron expuestos a infliximab en el útero, hasta 12 meses después del nacimiento (Julsgaard et al., 2016). Estos lactantes pueden tener un mayor riesgo de infección, inclu- yendo infecciones diseminadas graves que pueden llegar a ser mortales; es el caso, por ejemplo, de la infección diseminada por el bacilo de Calmette y Guérin (BCG), que ha sido notificada después de la administración de la vacuna de microorganismos vivos BCG tras el nacimiento.
Por tanto, se recomienda un periodo de espera de 12 meses después del nacimiento, antes de la administración de vacunas de microorganismos vivos a lactantes que estuvieron expuestos a infliximab en el útero. Cuando se prevé un beneficio clínico evidente para el lactante, se podría considerar la administración de una vacuna de microorganismos vivos en una etapa más temprana si los niveles séricos de infliximab en el lactante son indetectables o si la administración de infliximab se limitó al primer trimestre del embarazo (cuando la transferencia placentaria de IgG se considera mínima).
Con respecto a la administración de vacunas de microorganismos vivos a lactantes que estuvieron expuestos a infliximab durante la lactancia, en la literatura se han publicado datos limitados que sugieren que se detectan niveles bajos de infliximab en la leche materna, en concentraciones de hasta el 5% del nivel sérico materno (Fritzsche et al., 2012). También se ha detectado infliximab en el suero de lactantes tras su exposición a infliximab a través de la leche materna. Se espera que la exposición sistémica en un lactante sea baja porque el fármaco se degrada principalmente en el tracto gastrointestinal.
En base a estos datos, las recomendaciones europeas acordadas en el seno del comité PRAC son las siguientes: