Número 451, Marzo 2022




Además de la información que se incluye en los listados mensuales publicados en PAM, en BOT PLUS se incluye un apartado de Histórico, en las fichas de medicamentos, en el que se presenta información referente a cambios que haya sufrido anteriormente el medicamento o producto, entre otros, los cambios de nombre y los cambios de laboratorio. Esta información también está disponible para productos sanitarios financiados o dietoterápicos.
Se añade la posibilidad de visualización de las situaciones anteriores (o incluso futuras) relacionadas con un cambio de nombre. Con automatismos que nos permiten localizar un medicamento que haya cambiado de nombre, independientemente de cuál usemos.

Además de la información existente en Histórico, se permite la explotación de la información incluida en BOT PLUS en este apartado, mediante la integración de la información almacenada en Histórico en el apartado de Listados de BOT PLUS, que permite realizar consultas entre rangos de fechas y por un concepto en concreto de entre los almacenados en el apartado de Histórico. Entre ellos se incluyen, precisamente, los conceptos “Cambio del nombre del medicamento” y “Cambio del laboratorio comercializador”.

Resumen de las notas sobre seguridad y farmacovigilancia publicadas por la AEMPS desde principios del año 2021. Para información más ampliada y acceso al documento de la AEMPS, puede consultar BOT PLUS.
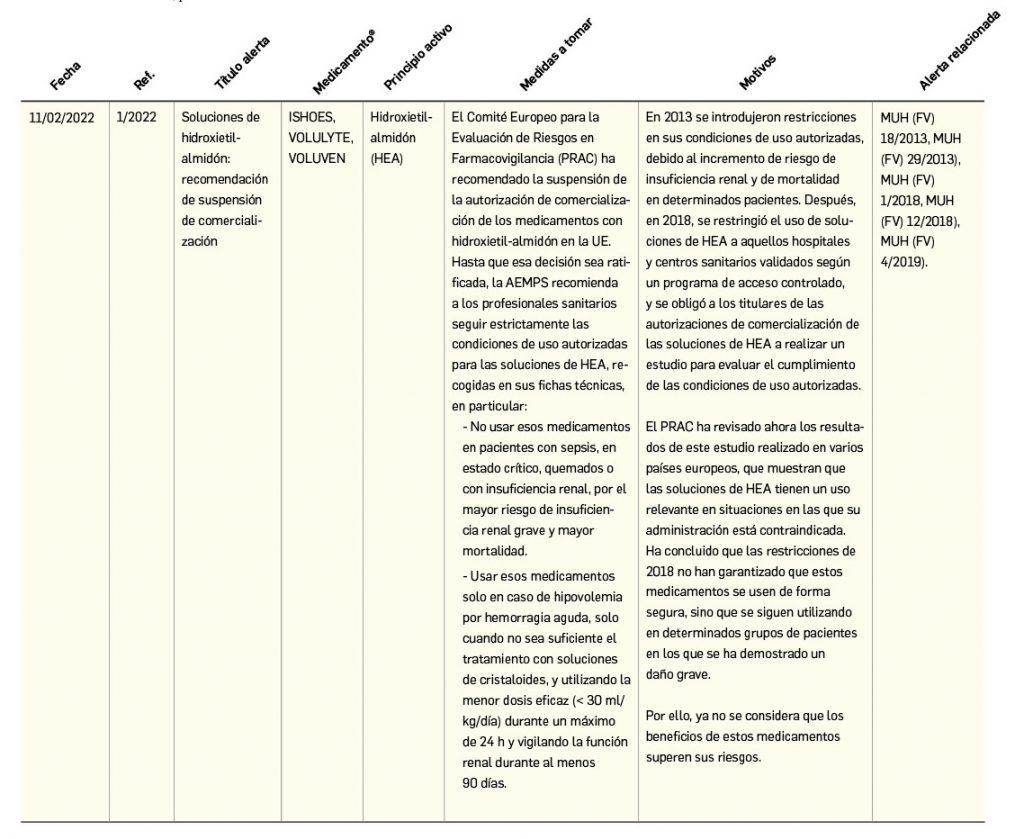
Alertas debidas a defectos de calidad observados en medicamentos de uso humano, publicadas por la AEMPS desde el anterior número y que suponen la retirada o inmovilización de ciertos lotes de medicamentos. En BOT PLUS puede encontrar más información detallada, con acceso al documento de la AEMPS.
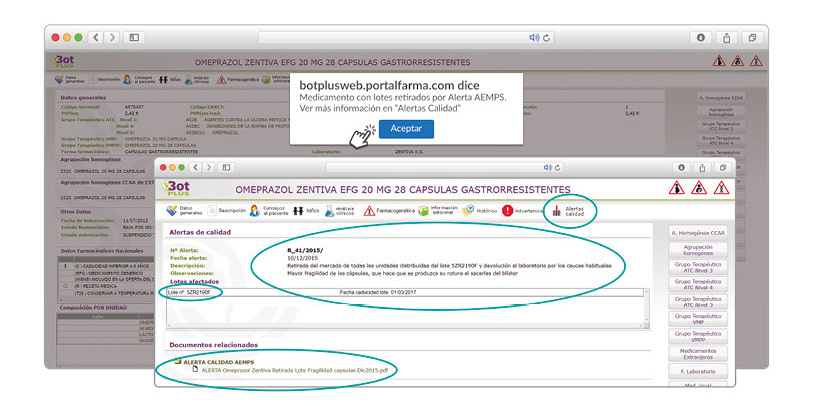
Además de los listados mensuales que podemos consultar en PAM, en BOT PLUS se incorpora la información que publica la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) relativa a notificaciones sobre seguridad y/o calidad de los medicamentos. Mediante un pictograma específico se pueden visualizar de forma rápida medicamentos afectados por alguna alerta de seguridad o de defectos de calidad, con tan solo entrar en su ficha.
Al acceder a la ficha de un medicamento afectado por una retirada, se visualiza un mensaje con la advertencia “Medicamento con lotes retirados por Alerta AEMPS. Ver más información en “Alertas Calidad”. Además, se incluye una pestaña específica en la que se pueden consultar los lotes concretos que han sido retirados, con sus respectivas fechas de caducidad, así como la descripción del defecto de calidad detectado y las medidas a adoptar. También se cuenta con acceso al documento publicado por la AEMPS.
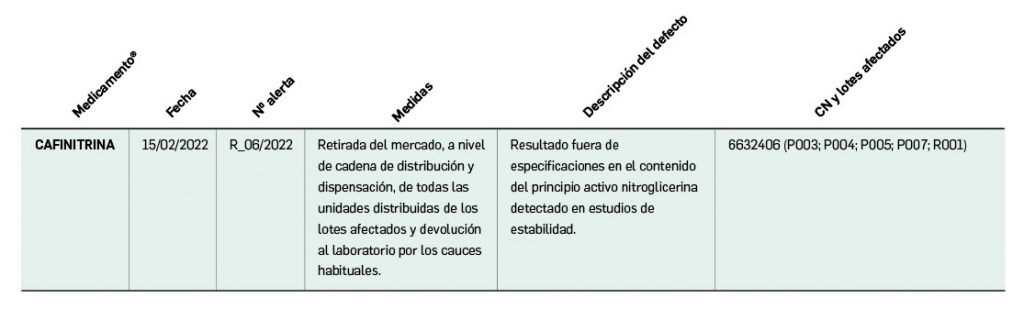
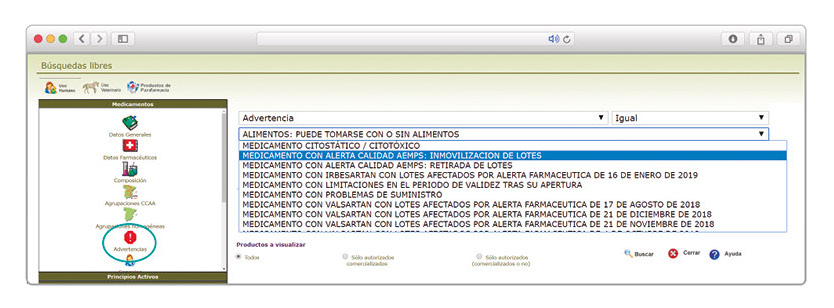
De forma interesante, dicha información se puede explotar a través de la Búsqueda Libre de BOT PLUS para obtener listados de todos los medicamentos afectados por alertas de calidad que implica la retirada (o también la inmovilización) de sus lotes en un momento dado.
Esta codificación de los lotes retirados es una información puesta a disposición de todos los usuarios, con el objetivo de ofrecer una nueva información capaz de integrarse con otros sistemas de información y mejorar la gestión e identificación de estos medicamentos, en los que la labor asistencial y de control del farmacéutico es fundamental.
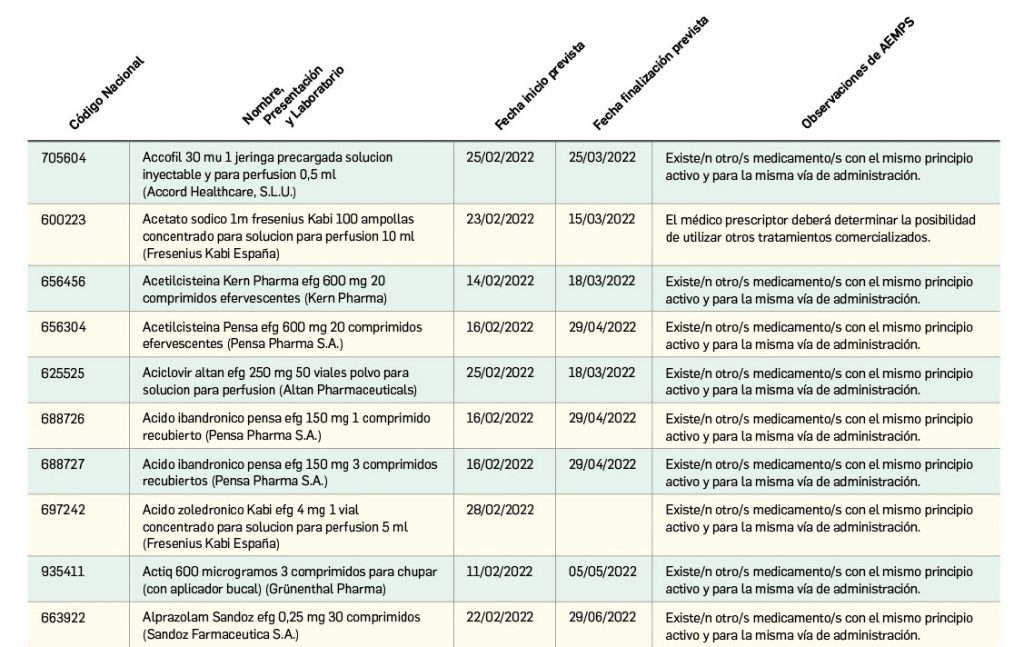
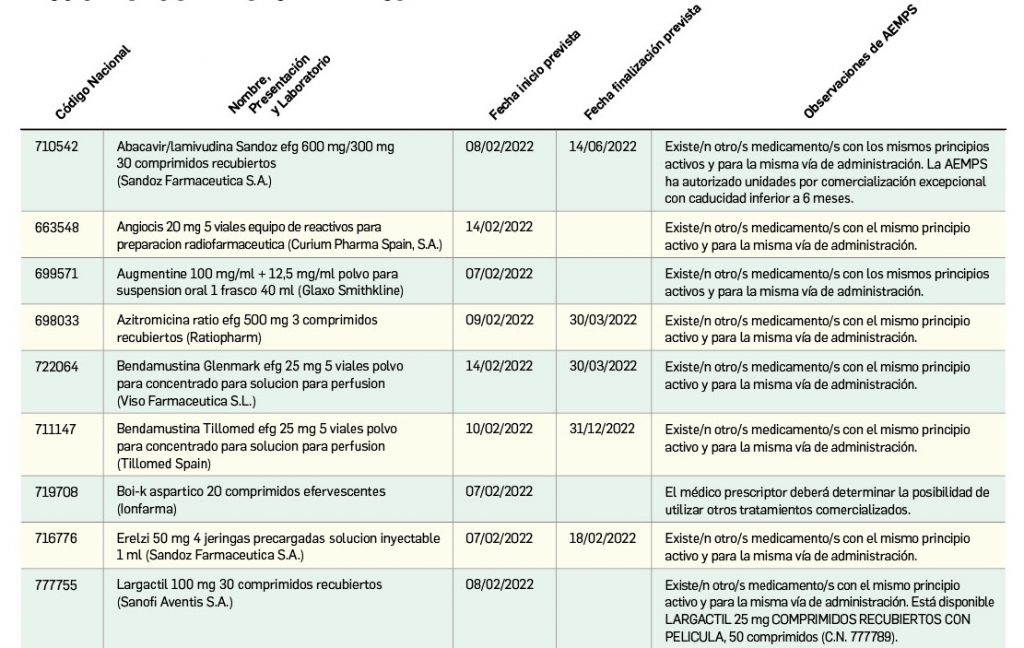
Listado de medicamentos con problemas de suministro publicado por la AEMPS, a fecha de cierre de este número. En BOT PLUS, se puede encontrar la información completamente actualizada, al tratarse de una información que varía de forma continua.
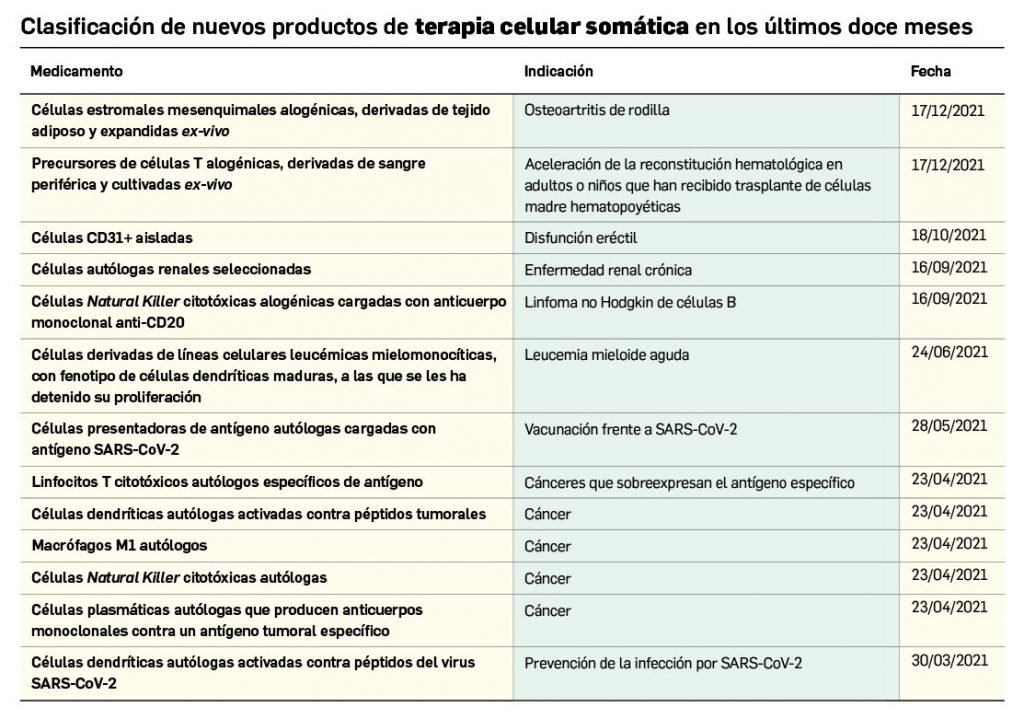
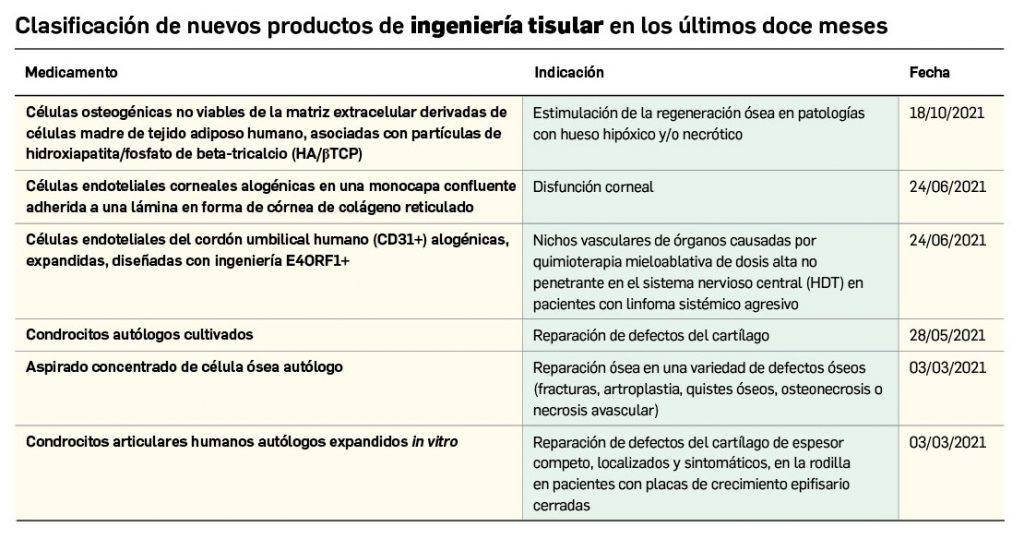
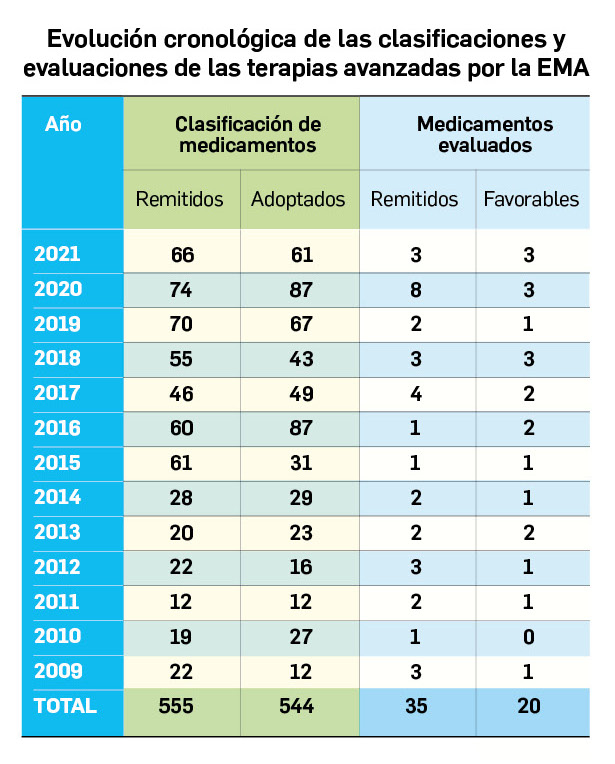
Los medicamentos de terapia avanzada (MTA o Advanced Therapy Medicinal Products, ATMP) ofrecen nuevos e innovadores tratamientos para las enfermedades. Están basados en la terapia génica, la terapia celular somática o la ingeniería tisular. El marco legal para las ATMP en la Unión Europea está establecido en la Regulation (EC) No 1394/2007 on advanced therapy medicinal products que asegura el libre movimiento de estas medicinas dentro de la Unión Europea y el acceso a los mercados. La regulación (EC) nº 1394/2007 también establece el nuevo Comité en Terapias avanzadas (CAT) cuya responsabilidad fundamental consiste en preparar un proyecto de opinión sobre cada nueva solicitud de medicamento de terapia avanzada planteada a la Agencia Europea de Medicamentos, antes de que el Comité de Medicamentos de Uso Humano (CHMP, Committee for Medicinal Products for Human Use) de la misma adopte una opinión definitiva sobre la concesión, modificación, suspensión o revocación de una autorización de comercialización para el medicamento en cuestión.
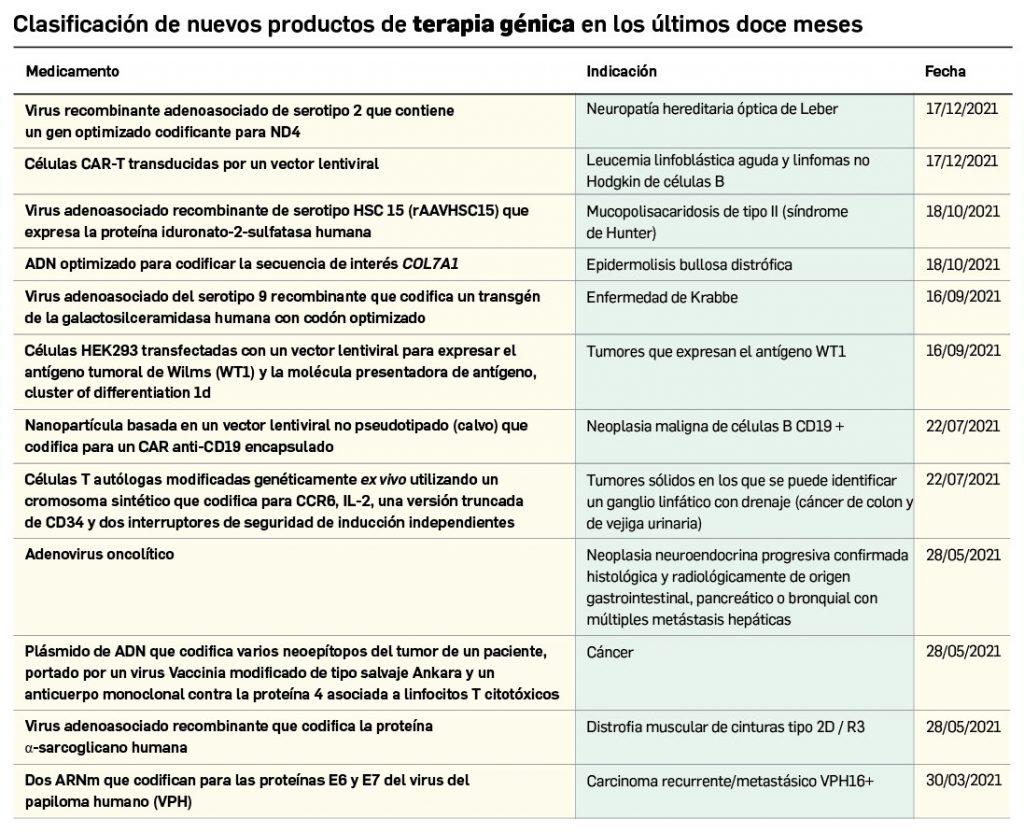
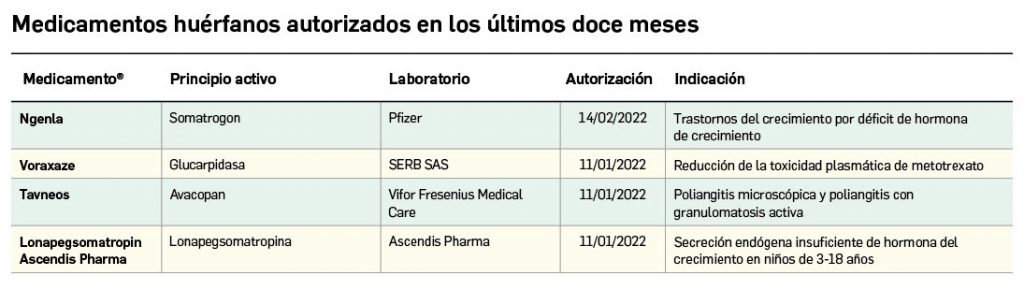
Los medicamentos huérfanos son aquéllos que sirven para diagnosticar, prevenir o tratar enfermedades raras de carácter muy grave o con riesgo para la vida. En la Unión Europea, la calificación de enfermedad rara se aplica a todas aquellas que no afectan a más de 5 de cada 10.000 personas. La designación de un medicamento como huérfano no garantiza su uso en la condición designada y no implica necesariamente que el producto satisfaga los criterios de eficacia, seguridad y calidad necesarios para la concesión de la autorización de comercialización. Como para cualquier medicamento, estos criterios solo pueden ser evaluados una vez que la solicitud de autorización de comercialización haya sido presentada.
► Instituto de Salud Carlos III (Ministerio de Ciencia e Innovación):
Instituto de Investigación en Enfermedades Raras:
https://www.isciii.es/QuienesSomos/CentrosPropios/IIER/Paginas/default.aspx
CIBERER (Centro de Investigación Biomédica en Red de Enfermedades Raras): https://www.ciberer.es/
► Instituto de Mayores y Servicios Sociales (IMSERSO, Ministerio de Derechos Sociales y Agenda 2030):
http://www.imserso.es/imserso_01/index.htm
► Federación Española de Enfermedades Raras (FEDER): www.enfermedades-raras.org
► Asociaciones de pacientes en España: https://enfermedades-raras.org/index.php/asociaciones/nuestros-socios
► Agencia Europea de Medicamentos (EMA; Europea Medicines Agency). Apartado de Medicamentos Huérfanos
(inglés): https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview
https://www.ema.europa.eu/en/committees/committee-orphan-medicinal-products-comp
► Comisión Europea: web oficial de la Comisión Europea sobre enfermedades raras y medicamentos huérfanos (español).
http://ec.europa.eu/health/rare_diseases/policy/index_es.htm
► Orphanet: Portal de información oficial de la Unión Europea sobre enfermedades raras y medicamentos huérfanos (español).
http://www.orpha.net/consor/cgi-bin/index.php?lng=ES
► Eurordis: Federación Europea de Asociaciones de Pacientes con Enfermedades Raras (español). http://www.eurordis.org/es
► Food & Drug Administration (FDA, Estados Unidos). Apartado de Medicamentos Huérfanos (inglés):
http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/default.htm
► Pharmaceuticals & Medical Devices Agency. Agencia de Medicamentos y Dispositivos Médicos, de Japón (inglés):
http://www.pmda.go.jp/english/index.html
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha remitido a los profesionales sanitarios, a través del laboratorio titular del medicamento con cladribina (Mavenclad®), la información reciente con recomendaciones para prevenir daño hepático grave. Así, antes de iniciar el tratamiento, se debe de realizar una revisión detallada de la historia clínica sobre enfermedades hepáticas subyacentes o episodios de daño hepático con otros medicamentos; también se deberán realizar pruebas de función hepática que incluyan niveles de transaminasas, fosfatasa alcalina y bilirrubina total al inicio de cada año de tratamiento. Durante el tratamiento, se deben llevar a cabo pruebas de función hepática tantas veces como se considere clínicamente necesario. Y, en caso de que un paciente desarrolle alteración hepática, el tratamiento debe interrumpirse.
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha remitido información (AEMPS, 2022) a través del laboratorio titular de la autorización de comercialización del medicamento con cladribina (Mavenclad®) mediante una comunicación directa a profesionales sanitarios sobre nuevos datos de seguridad (como DHPC, según siglas del inglés). La misma comunicación la han realizado todas y cada una de las 27 agencias reguladoras nacionales europeas (por ejemplo, HPRA, 2022).
La cladribina es un fármaco indicado para el tratamiento de pacientes adultos con esclerosis múltiple (EM) recurrente muy activa definida mediante características clínicas o de imagen. Durante el tratamiento con Mavenclad®, se han notificado casos de daño hepático, incluidos casos graves y casos que han conducido a la suspensión del tratamiento y la revisión de los datos de seguridad disponibles concluyen que el tratamiento incrementa el riesgo de daño hepático.
En la mayoría de los casos de alteración hepática se acompañaron de síntomas clínicos leves. Sin embargo, en casos raros, se produjo ictericia y una elevación transitoria de las transaminasas superior a 1.000 U/l. El tiempo transcurrido desde el inicio del tratamiento hasta la aparición de la reacción fue variable, aunque la mayoría de los casos ocurrieron dentro de las 8 semanas posteriores al primer ciclo de tratamiento. En la revisión de los casos no se identificó un mecanismo claro. Algunos pacientes tenían antecedentes de episodios previos de daño hepático con otros medicamentos o trastornos hepáticos subyacentes. Los datos de los ensayos clínicos no sugieren un efecto dosis-dependiente.
Se ha actualizado la información del medicamento (ficha técnica y prospecto) para incluir la “alteración hepática” como una reacción adversa poco frecuente del medicamento. Además, se han incorporado nuevas advertencias y precauciones incluyendo las recomendaciones de realizar una anamnesis adecuada y de evaluar las pruebas de función hepática antes del inicio de cada año de tratamiento. Por último, se actualizarán los materiales sobre prevención de riesgos, incluyendo la guía del prescriptor, y la guía del paciente con el objeto de reflejar estas nuevas recomendaciones.
Con el fin de evitar los casos de daño hepático grave, se hacen las siguientes recomendaciones:
Es importante informar a los pacientes sobre este riesgo y de que deben llevar a cabo las revisiones y las pruebas de función hepática al inicio de cada año de tratamiento, y durante el tratamiento, según el criterio del médico prescriptor. Además, se debe aportar la recomendación final de que busquen atención médica si presentan cualquier signo o síntoma sugestivo de lesión hepática.