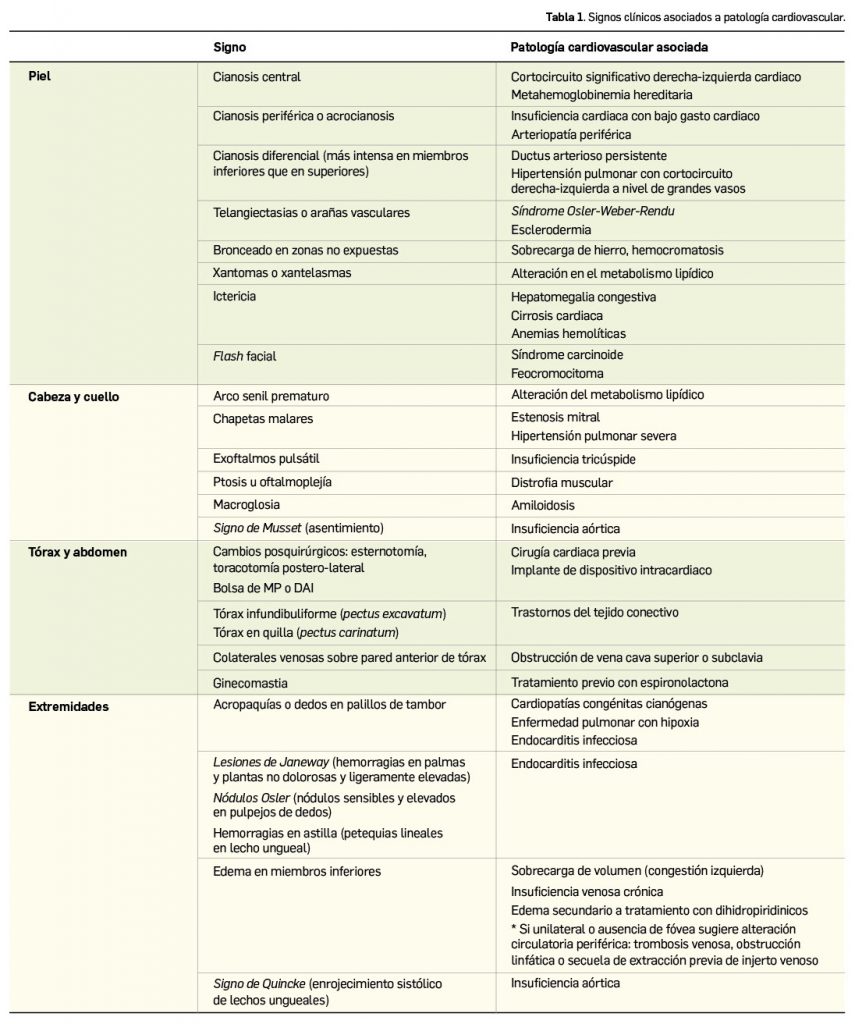
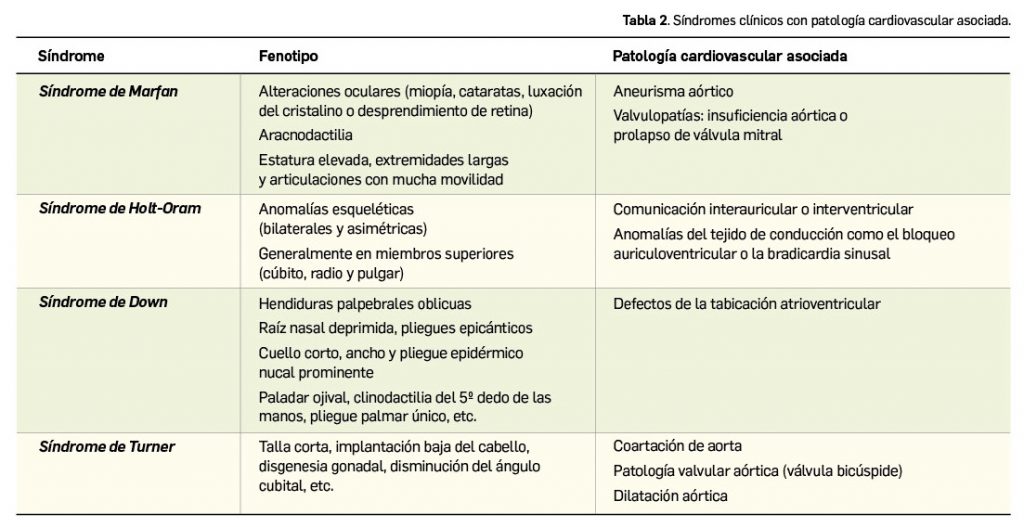
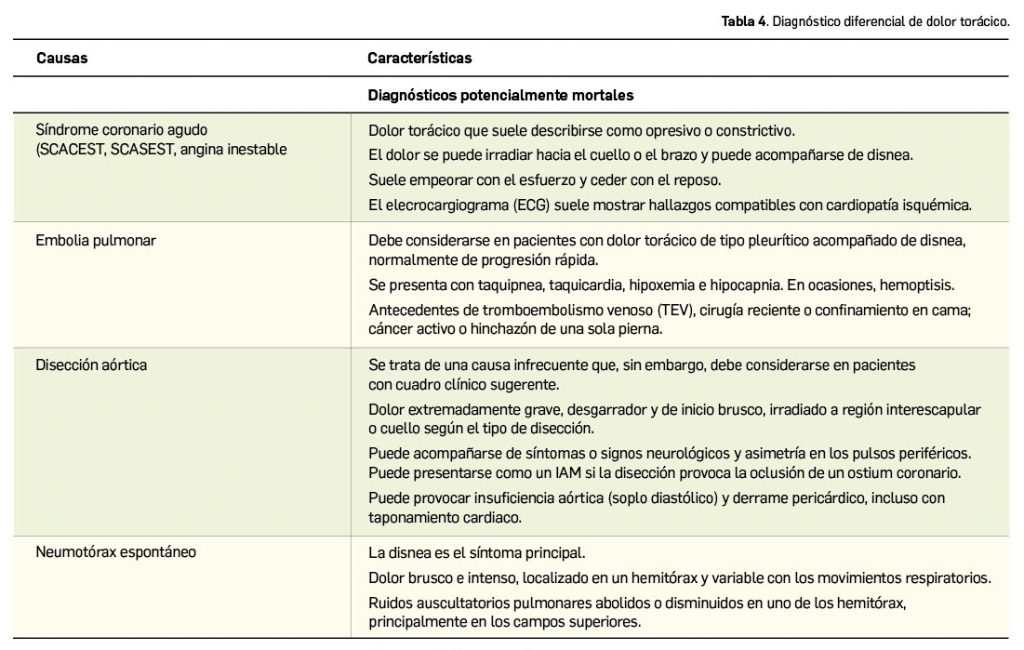
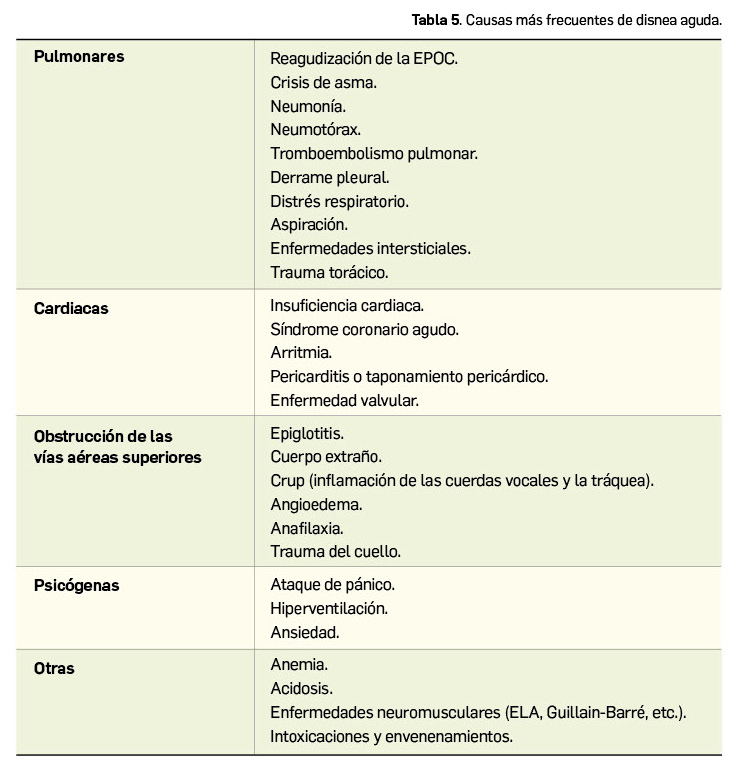
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha remitido recientemente a los profesionales sanitarios, a través de los laboratorios titulares de los medicamentos con anagrelida (Xagrid® y EFG), la información sobre el mayor riesgo de complicaciones trombóticas, incluido el infarto cerebral, tras la interrupción brusca de tratamiento de anagrelida. En base a ello, se recomienda evitar la interrupción brusca de este tratamiento debido al riesgo de aumento repentino en el recuento de plaquetas y, como consecuencia, un aumento de complicaciones trombóticas potencialmente mortales, como por ejemplo el infarto cerebral.
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha remitido información (AEMPS, 2022) a través de los laboratorios titulares de la comercialización de los medicamentos con anagrelida (Xagrid® y EFG) mediante una comunicación directa a profesionales sanitarios sobre nuevos datos de seguridad (como DHPC, según siglas del inglés). Todas las 27 agencias reguladoras nacionales europeas han realizado la misma comunicación DHPC, como la agencia irlandesa (por ejemplo, HPRA, 2022).
El clorhidrato de anagrelida está indicado para la reducción del recuento elevado de plaquetas en pacientes de riesgo con trombocitemia esencial que no toleran el tratamiento que están siguiendo o cuyo recuento elevado de plaquetas no disminuye hasta un nivel aceptable con dicho tratamiento.
Como resultado de un análisis acumulativo de la base de datos de seguridad de la compañía realizado hasta el 6 de agosto de 2021, se revelaron 15 eventos de complicaciones trombóticas, incluido infarto cerebral, ocurridos después de una interrupción reciente de anagrelida. Se ha concluido que el infarto cerebral, junto con otras complicaciones trombóticas, si bien pueden asociarse a la condición/indicación preexistente, también pueden producirse tras la interrupción brusca de anagrelida, con una dosificación inadecuada o bien por la falta de eficacia.
El infarto cerebral posterior a la interrupción brusca de anagrelida puede presentarse por un mecanismo relacionado con un efecto rebote en el recuento de plaquetas. Normalmente comenzará a aumentar este rebote durante los 4 días siguientes a la interrupción del tratamiento, y volverá a niveles basales tras 1 o 2 semanas, posiblemente superando dichos valores basales.
Se actualizará la información de seguridad según la información disponible, tanto de la sección 4.4. “Advertencias y precauciones especiales de empleo” como de la sección 4.8. “Reacciones Adversas” de la ficha técnica de los medicamentos con anagrelida, para reflejar los últimos datos y recomendaciones.
Recomendaciones a los profesionales sanitarios
En base a la información generada con los nuevos datos, se establecen las siguientes recomendaciones:
- Se debe evitar la interrupción brusca de este tratamiento con anagrelida (Xagrid® y EFG) debido al riesgo de aumento repentino en el recuento de plaquetas y, como consecuencia, aumento de complicaciones trombóticas potencialmente mortales, como por ejemplo el infarto cerebral.
- En caso de interrupción de la dosis, o la retirada del tratamiento, se deberá realizar una monitorización frecuente del recuento de plaquetas (para más información consulte la sección 4.4 de la ficha técnica de anagrelida –Xagrid® y EFG).
- Los pacientes deben ser informados sobre cómo reconocer los signos y síntomas tempranos que sugieran complicaciones trombóticas, como un infarto cerebral, y acerca de la necesidad de buscar asistencia médica inmediata en caso de presentar dichos síntomas.
Recomendaciones a los pacientes
Los pacientes en tratamiento con anagrelida (Xagrid® y EFG) deben recibir información sobre cómo reconocer los signos y síntomas tempranos que sugieran complicaciones trombóticas, como un infarto cerebral, y sobre la necesidad que tienen de buscar asistencia médica inmediata en caso de presentar dichos síntomas.