Resumen
Acalabrutinib es un nuevo inhibidor potente y altamente selectivo de la tirosina cinasa de Bruton (BTK), una enzima que participa en la señalización bioquímica del receptor de antígenos y del receptor de citocinas de las células B, favoreciendo la supervivencia y proliferación de linfocitos B tumorales. Así, la inhibición covalente e irreversible que inducen acalabrutinib y su metabolito ACP-5862 sobre el centro activo de la enzima se traduce en una inhibición de la adhesión y migración dependiente de integrinas de linfocitos B, conducente a la reducción tumoral. El medicamento ha sido autorizado para el tratamiento por vía oral (100 mg/2 veces al día) en monoterapia o en combinación con obinutuzumab de pacientes adultos con leucemia linfocítica crónica (LLC) no tratados previamente, y para su uso en monoterapia en pacientes adultos con LLC que han recibido ≥ 1 tratamiento previo.
Su aprobación se sustentó en los datos clínicos de dos estudios pivotales aleatorizados y multicéntricos, de fase 3, abiertos, simple ciego y de grupos paralelos. En uno de ellos (ELEVATE-IN), con 535 pacientes adultos con LLC no tratados previamente, la asociación acalabrutinib+obinutuzumab y la monoterapia de acalabrutinib mostraron una superioridad notable frente a una combinación de obinutuzumab y clorambucilo (control activo). Tras 28 meses, sin alcanzarse la mediana de SLP en los brazos del fármaco (vs. 22,6 meses en el grupo control), la administración de acalabrutinib con o sin obinutuzumab redujo en un 90% y un 80% el riesgo de progresión o muerte por la enfermedad, respectivamente; se alcanzaron estimaciones de SLP a 24 meses del 93% y del 87%. El otro estudio (ASCEND) incluyó a pacientes adultos con LLC refractaria o en recaída tras ≥ 1 tratamiento previo (N= 310). Tras 16 meses, la monoterapia con acalabrutinib demostró una eficacia superior al régimen activo usado como control (idelalisib + rituximab o bendamustina + rituximab): tampoco se alcanzó la mediana de SLP en el brazo con el nuevo fármaco (vs. 16,5 meses), y se estimó una reducción del 69% en el riesgo de muerte o progresión. En general, la eficacia de acalabrutinib se reveló duradera y consistente en pacientes con factores citogenéticos de alto riesgo. Los datos de supervivencia global (aún inmaduros) y de tasa de respuesta reafirman la utilidad clínica del fármaco tanto en pacientes naïve como pretratados.
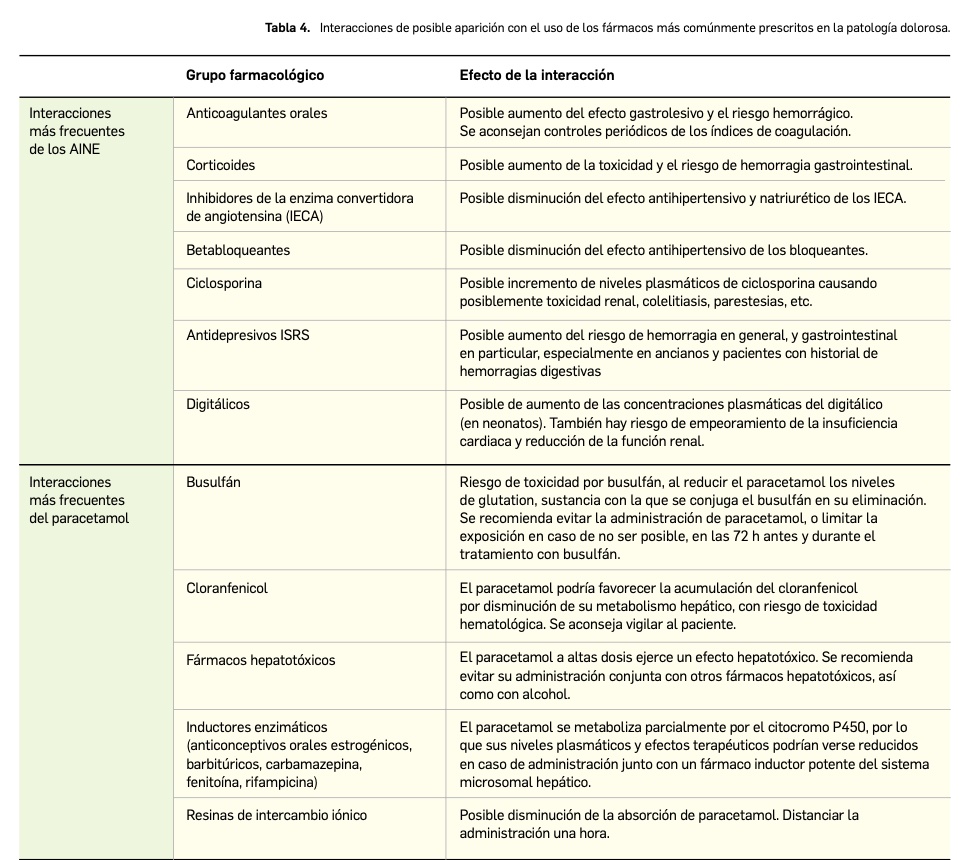
En términos de seguridad, tiene un perfil toxicológico notable, con frecuentes y relevantes eventos adversos en los dos tipos de pacientes. La incidencia de eventos adversos de intensidad de grado 3-4 fue del 50-70% y supone tasas de discontinuación del tratamiento del 10-11% y de reducción de dosis del 4-7%. Las reacciones adversas más frecuentes (> 20%) fueron infección, dolor de cabeza, diarrea, hematomas, dolor musculoesquelético, náuseas, fatiga, tos y erupción cutánea. Entre las más graves, sobresalen las infecciones y las citopenias; pero tampoco se puede obviar el riesgo aumentado de segundas neoplasias primarias. No se observaron diferencias relevantes de seguridad en pacientes de ≥ 65 años o en aquellos con perfil citogenético de peor pronóstico.
En definitiva, se trata de un inhibidor de BTK de 2ª generación cuya mayor selectividad por la enzima hace que sea al menos tan eficaz como ibrutinib pero con un perfil de toxicidad mejorado; presenta, por ejemplo, un menor riesgo de eventos adversos off target a nivel cardiaco (por ejemplo, de generación de arritmias). Sin embargo, no se dispone aún de comparaciones entre ambos, y habrá que esperar a un estudio ahora en marcha en pacientes con LLC en recaída/refractariedad y alto riesgo citogenético para confirmar sus diferencias en eficacia y seguridad. Por ahora, es probable que el uso de acalabrutinib, tanto en monoterapia como asociado a obinutuzumab, se considere una opción preferencial en primera línea para los pacientes con LLC no tratados, similar al uso de ibrutinib o venetoclax (mayoritariamente asociados a obinutuzumab). En pacientes con LLC pretratados la monoterapia con acalabrutinib viene a completar las posibilidades de tratamiento junto a ibrutinib o venetoclax más rituximab, aportando una respuesta clínica relevante en cuadros con escasas opciones terapéuticas, incluso cuando éstas no son funcionales. A la vista de lo demostrado para ibrutinib, es previsible que en un futuro se pruebe un beneficio clínico con acalabrutinib en otras neoplasias de células B.
Aspectos fisiopatológicos
La leucemia linfática crónica o leucemia linfocítica crónica (LLC) se considera un linfoma linfocítico o leucémico de bajo grado, caracterizado por la acumulación de linfocitos B maduros en la sangre, médula ósea y órganos linfáticos. Los linfocitos circulantes son morfológicamente similares a los normales, pero funcionalmente anormales: son células maduras pero más pequeñas1, con una estrecha separación entre el citoplasma y un núcleo denso que carece de nucleolo distinguible y tiene la cromatina parcialmente agregada; expresan marcadores de superficie CD5, CD20 y CD23. La acumulación se inicia frecuentemente en la médula ósea, diseminándose posteriormente hacia los ganglios linfáticos y bazo, pudiendo detectarse esplenomegalia.
Es la leucemia más frecuente en los países occidentales, sobre todo en Europa y Norte América, constituyendo el 30% de todas las formas de leucemia y 75% de las leucemias crónicas. Su incidencia media en la Unión Europea es de unos 4-5 casos/100.000 habitantes/año, más común en personas de mayor edad (mediana de edad al diagnóstico de 70 años), siendo rara antes de los 40 años. A la edad de 50 años alcanza los 5 casos nuevos/100.000 habitantes/año y a los 80 años llega a los 30 casos/100.000 habitantes/año. Existe un predominio en el sexo masculino (2:1), y afecta a más de 300.000 personas en el mundo. En España, se estima que afecta a más de 15.000 personas; concretamente, se diagnostican alrededor de 1.800 nuevos casos cada año (el 90% en pacientes de > 50 años y muy pocos casos se diagnostican por debajo de los 30 años). Es previsible que la prevalencia de la enfermedad aumente con el paso de los años y el envejecimiento de la población, dada la alta expectativa de vida en nuestro país (82,8 años de media en ambos sexos según datos de la OMS en 2015).
Como se ha indicado, los pacientes con LLC activa se caracterizan por una acumulación progresiva de linfocitos B (el diagnóstico requiere la presencia de al menos 5.000 linfocitos B en sangre periférica durante al menos 3 meses), a veces con linfadenopatía, hepatoesplenomegalia, anemia y trombocitopenia. Asimismo, produce un estado de inmunosupresión que incrementa el riesgo de infecciones, que en última instancia es la principal causa de muerte en estos pacientes. Además del subtipo más frecuente de LLC –el que afecta a células B, representando más del 97% de los casos–, se identifican un 2-3% de casos en que la proliferación clonal anormal se produce a partir de células T. E incluso también se han incluido otros patrones leucémicos crónicos dentro de la LLC, tales como la leucemia prolinfocítica, la fase leucémica del linfoma cutáneo de células T (síndrome de Sézary), la leucemia de células peludas (tricoleucemia) y el linfoma leucemizado.
Sea como fuere, el origen de la LLC sigue siendo desconocido, aunque se apuntan varias hipótesis, como el efecto de radiaciones ionizantes, los agentes alquilantes o ciertos productos leucemógenos, que parecen aumentar el riesgo de su desarrollo. La acumulación de linfocitos parece deberse a un funcionamiento erróneo en la apoptosis (muerte celular programada); no obstante, se han descrito otros mecanismos que posiblemente colaboren de alguna manera en la acción proliferativa, como ciertas interleucinas (IL) o sus receptores, como el factor de necrosis tumoral (TNF) o las IL-4 e IL-6. Aproximadamente la mitad de los pacientes, y aún más en estadios avanzados, presentan algún tipo de alteración citogenética.
Desde el punto de vista clínico, la patología tiene una amplia variabilidad de presentaciones y comportamiento, desde una mayoría de pacientes que viven muchos años con linfocitosis asintomática (sin requerir tratamiento) a otros que requieren tratamiento precozmente, con breves respuestas, y fallecen a causa de su enfermedad en pocos años (AEMPS, 2017). En consonancia, la mediana de supervivencia desde el diagnóstico oscila dependiendo de la presencia de factores de riesgo: en pacientes asintomáticos en estadios iniciales, la supervivencia suele ser superior a 10 años, mientras que, en pacientes con enfermedad avanzada, sintomática o progresiva, la mediana de supervivencia varía entre 18 meses y 3 años. Además, los pacientes con LLC también son más propensos a desarrollar una segunda neoplasia.
Cabe destacar que se han descrito tres grupos pronósticos en función de la citogenética, siendo peor para los casos relacionados con una mutación TP53, una translocación t(11q;v) o una deleción del(11q) o del(17p)2; particularmente, esta última, junto con la mutación TP53, confiere resistencia a la inmunoquimioterapia (sobre todo, a fludarabina) y se considera como de muy alto riesgo. En ambos casos la mediana de supervivencia es de 2-3 años y, aunque son casos relativamente infrecuentes en el diagnóstico inicial (7% para del 17p y 8-12% para TP53), suponen prácticamente el 50% de los casos recidivantes de LLC. La trisomía del par cromosómico 12 (+12), la alteración citogenética más frecuente (25-30%), se asocia con un pronóstico de gravedad intermedia, mientras que los casos con mejor pronóstico son aquellos cuya anomalía citogenética implica una deleción del(13q).
Desde el punto de vista del tratamiento, la mayoría de los pacientes que presentan una linfocitosis asintomática no precisan tratamiento, en cuyo caso se recomienda un seguimiento clínico periódico. Sin embargo, el tratamiento es necesario en pacientes con enfermedad avanzada (con anemia o trombopenia), en progresión o sintomática, con alta carga tumoral, presencia de síntomas B o infecciones de repetición. En ninguno de los casos la terapia es curativa, con excepción del trasplante alogénico de progenitores hematopoyéticos (TPH), para el que una mayoría de pacientes no se consideran aptos. E incluso algunos expertos plantean que el sobretratamiento puede tener más efectos deletéreos sobre el paciente que el infratratamiento.
La farmacoterapia específica incluye quimioterapia citotóxica (fludarabina, ciclofosfamida) y e inmunoquimioterapia –con anticuerpos anti-CD-20 como rituximab, ofatumumab y obinutuzumab–, corticoides y radioterapia. La cirugía (TPH) o la radioterapia solo son útiles en casos concretos. Los tres últimos fármacos específicamente autorizados para esta indicación han sido: ibrutinib, idelalisib y venetoclax.
El ibrutinib actúa inhibiendo de forma irreversible y selectiva a la tirosina cinasa de Bruton (BTK), un miembro de la familia de las tirosina cinasas Tec que participa en la señalización bioquímica del receptor de antígenos (BCR) y del receptor de citocinas de los linfocitos B, implicados en la patogenia de diversas neoplasias de linfocitos B; dicha inhibición impide la adhesión y migración dependientes de integrinas de los linfocitos B. Fue inicialmente autorizado, como medicamento huérfano, para el tratamiento por vía oral de pacientes adultos con LLC que han recibido al menos un tratamiento previo, o en primera línea en presencia de deleción del 17p o mutación de TP53 en pacientes en los que la inmunoquimioterapia no se considera apropiada. El tratamiento se asocia con altas tasas de respuesta a largo plazo en pacientes con LLC recidivante/refractaria y linfoma de células del manto, incluidos pacientes con lesiones genéticas de alto riesgo. Esto, unido a la facilidad de posología hace que ibrutinib sea hoy en día un tratamiento ampliamente utilizado para pacientes con LLC tanto en primera línea como en recaída o refractariedad.
Por su parte, el idelalisib, un inhibidor selectivo de la fosfatidilinositol 3-cinasa p110δ (PI3Kδ) –mecanismo por el cual inhibe la acción del receptor de células B (BCR)– ha sido autorizado para el tratamiento, en combinación con rituximab, de los pacientes adultos con LLC que han recibido al menos un tratamiento anterior o como tratamiento de primera línea en presencia de la deleción del 17p o de la mutación de TP53 en pacientes no adecuados para quimioinmunoterapia. Los datos clínicos disponibles indican, en relación con un placebo y siempre en asociación a rituximab, una notable superioridad, tanto en términos de supervivencia libre de progresión tumoral (SLP) como de supervivencia global (SG) y tasa de respuesta objetiva (75 vs 15%); una superioridad manifiesta incluso en los pacientes con mutaciones del17p y/o TP53, como con IGHV (fragmento variable de las cadenas pesadas de las inmunoglobulinas) no mutado y en personas con ≥ 65 años.
El último fármaco incorporado para el tratamiento de la LLC ha sido venetoclax, un inhibidor potente y selectivo de la proteína antiapoptótica Bcl-2, que es sobreexpresada por las células de la LLC y en otros tipos celulares tumorales. El medicamento, autorizado como huérfano, está indicado para el tratamiento en monoterapia de la LLC en presencia de deleción 17p o mutación del gen TP53 en pacientes adultos que no son adecuados o han fallado al tratamiento con un inhibidor de la vía del receptor de antígenos del linfocito B; también está indicado en monoterapia para el tratamiento de la LLC en ausencia de dichas mutaciones en adultos que han fallado al tratamiento con inmunoquimioterapia y a un inhibidor de la vía del receptor de antígenos del linfocito B. Venetoclax inauguró una nueva vía farmacológica en oncología, potencialmente útil en cuadros resistentes o refractarios de LLC y en algunas otras formas de leucemia ligadas a linfocitos B, aportando una respuesta clínica relevante en cuadros con muy escasas opciones terapéuticas, incluso cuando éstas no son funcionales (Cuéllar, 2018). Su uso, que se prefiere en tratamientos limitados en el tiempo (que tendría una teórica ventaja asociada al menor riesgo de efectos adversos, de aparición de resistencias y, quizás, mejor adherencia por los pacientes), requiere conocer la posibilidad de aparición de lisis tumoral, sobre todo en las fases iniciales del tratamiento y la toxicidad hematológica, fundamentalmente neutropenia, que puede producir.
En cualquier caso, si bien el mayor progreso se ha realizado en el campo de la identificación de marcadores moleculares y celulares que permiten predecir la evolución de la LLC (en mayor medida que en su terapéutica), las últimas guías de práctica clínica (NCCN, ESMO) reflejan que el inicio y la selección del tratamiento no debe basarse en los factores pronósticos, con la excepción de pacientes que presenten las mutaciones del 17p y/o TP533(AEMPS, 2017). Se acepta que no existe un único tratamiento de primera línea adecuado para todos los pacientes debiendo de individualizarse para cada caso: la elección e inicio del tratamiento en la amplia mayoría de casos se establece fundamentalmente en base a la edad y fragilidad del paciente, según su capacidad de tolerar o no tratamiento con análogos de purinas.
En los últimos años se han desarrollado nuevos fármacos que pretenden cubrir las necesidades médicas no cubiertas en el tratamiento de los pacientes con LLC. Se han investigado nuevos inhibidores de PI3Kδ, como duvelisib o umbralisib, e inhibidores de BTK más específicos (acalabrutinib, zanubrutinib y tirabrutinib entre los inhibidores covalentes o LOXO-305 y vecabrutinib entre los no covalentes). Pero todavía la LLC es una enfermedad incurable y en la que, en ocasiones, deben realizarse diferentes tratamientos en las recaídas que se producen, constituyendo uno de los ejemplos de neoplasias hematológicas en la que los nuevos tratamientos que van apareciendo –basados en pequeñas moléculas orales junto con anticuerpos monoclonales anti-CD20– son la base del abordaje (a excepción de pacientes jóvenes sin citogenética adversa y patrón mutado de IGHV, en el que la combinación fludarabina, ciclofosfamida y rituximab puede considerarse al mismo nivel que el tratamiento con ibrutinib).
Acción y mecanismo
Acalabrutinib es un nuevo agente antineoplásico que actúa como inhibidor covalente y altamente selectivo de la tirosina cinasa de Bruton (BTK), de segunda generación, la cual participa en la señalización bioquímica del receptor de antígenos (BCR) y del receptor de citocinas de los linfocitos B, implicados ambos en la patogenia de diversas neoplasias de linfocitos B. El medicamento ha sido autorizado para el tratamiento por vía oral (100 mg/2 veces al día) en monoterapia o en combinación con obinutuzumab de pacientes adultos con leucemia linfocítica crónica (LLC) no tratados previamente, y también para su uso en monoterapia en pacientes adultos con LLC que han recibido al menos un tratamiento previo.
La BTK, como molécula implicada en la vía de señalización del BCR y del receptor de citocinas, favorece con su actividad la supervivencia y la proliferación de los linfocitos B, siendo clave en la adhesión y la quimiotaxis celular. Mediante su inhibición, acalabrutinib impide la adhesión y migración dependiente de integrinas de los linfocitos B. Es preciso citar que parte de la actividad del fármaco es mediada por su metabolito activo ACP-5862, que, como la molécula primaria, se unen específicamente en el hueco enzimático para el ATP de la enzima BTK, formando un enlace covalente con un resto de cisteína (Cys-481), lo cual determina el carácter irreversible de la inhibición: esto explica que, si bien acalabrutinib tiene una vida media corta tras su rápida absorción, la respuesta farmacodinámica se extienda en el tiempo. Durante su desarrollo clínico, se probó que, en pacientes oncológicos con neoplasias de células B, la mediana de ocupación de la BTK en estado de equilibrio se mantiene ≥ 95% en sangre periférica durante 12 h, lo cual asegura la inactivación de la enzima durante todo el intervalo de administración recomendado.
Los estudios preclínicos in vitro e in vivo permitieron demostrar que acalabrutinib (IC50= 3-5 nM) y su metabolito activo ACP-5862 (IC50= 5 nM) ejercen un efecto inhibitorio similar sobre la BTK, y muy escaso frente a la cinasa de la tirosin-proteína (TEC), del receptor del factor de crecimiento epidérmico (EGFR) y de la cinasa de la célula T inducible por la interleucina-2 (ITK), lo cual podría explicar el mejor perfil de seguridad frente a ibrutinib.
Además, los estudios clínicos pusieron de manifiesto que, con uso del fármaco en pacientes con LLC, a los 6 meses se produce un incremento de los linfocitos T CD8+ productores de interferón-γ (lo que puede contrarrestar el agotamiento de células T que se produce conforme progresa la LLC), así como una disminución de la expresión de PD-1, variables ambas relacionadas con la mejoría de la inmunidad T; respecto a la inmunidad humoral, se observó un aumento del nivel de IgA, que tiende a normalizarse, aunque no de IgG e IgM (EMA, 2020).
Aspectos moleculares
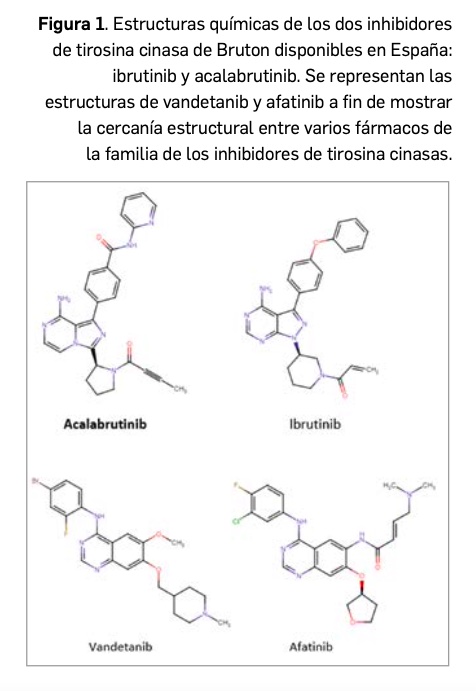
Acalabrutinib es un inhibidor de tirosina cinasa de Bruton (BTK) de segunda generación que tiene por nombre químico el de (S)-4-(8-amino-3-(1-but-2-inoilpirrolidin-2-il)-imidazo[1,5-α]pirazin-1-il)-N-(piridin-2-il)-benzamida; se corresponde con una fórmula molecular de C26H23N7O2 y un peso molecular de 465,51 g/mol. El principio activo contiene un centro quiral (de hecho, es el enantiómero S), se presenta como un polvo no higroscópico de color blanco-amarillo, y exhibe una solubilidad dependiente de pH en medio acuoso. Se han identificado varios polimorfos metaestables, tanto anhidros como hidratos y solvatos.
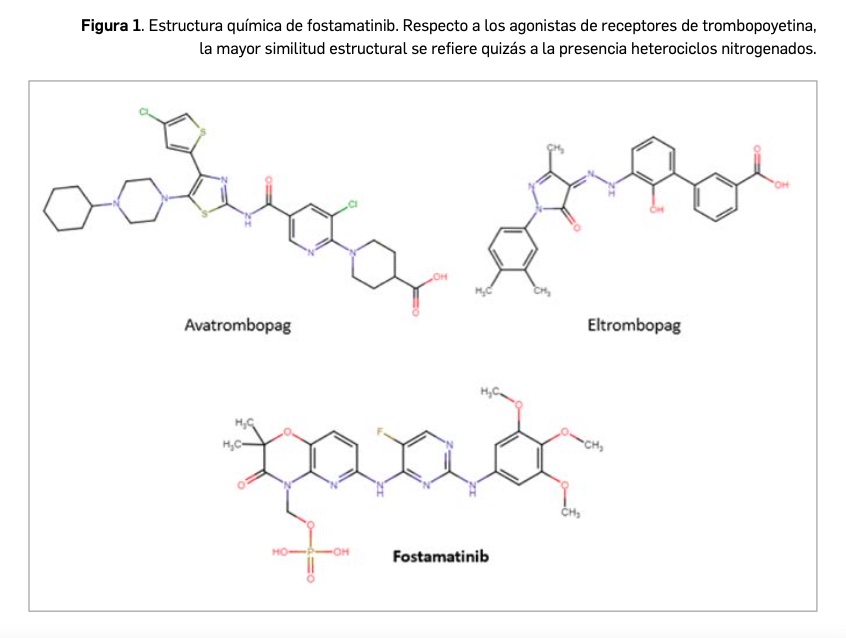
De forma similar a ibrutinib, con el que comparte una estrecha similitud estructural (Figura 1), se relaciona farmacológicamente también con afatinib y vandetanib. Es decir, se pueden vislumbrar ciertas características estructurales compartidas con otros miembros de la amplia serie de inhibidores de proteína cinasas, los cuales son el resultado de la optimización funcional mediante modelización molecular a partir de una serie de 2-fenilaminopirimidinas, de donde surgió imatinib, cabeza de serie del grupo. Todos ellos guardan –en mayor o menor grado– una familiaridad química con la molécula de ATP (o, en su caso, con la de GTP, como sucede en las cinasas MAPK), con la que compiten para provocar el bloqueo de la cinasa correspondiente.
Eficacia y seguridad clínicas
La autorización de acalabrutinib para su uso por vía oral en pacientes con LLC naïve o no tratados previamente se sustentó en los resultados de un estudio pivotal de fase 3 (ELEVATE-TN), multicéntrico y multinacional (142 centros de 18 países), con diseño abierto simple ciego y de grupos paralelos, que aleatorizó (1:1:1) a un total de 535 pacientes adultos4 a recibir un tratamiento de acalabrutinib (100 mg/12 h) con obinutuzumab, una monoterapia de acalabrutinib, o una combinación de obinutuzumab y clorambucilo por vía oral. Los tratamientos fueron administrados en ciclos de 28 días, manteniendo el nuevo fármaco hasta progresión de la enfermedad o toxicidad inaceptable; obinutuzumab y clorambucilo5 se administraron durante un máximo de 6 ciclos. Tras la progresión de la enfermedad, 45 pacientes aleatorizados inicialmente al brazo de obinutuzumab y clorambucilo cruzaron a recibir monoterapia con acalabrutinib.
El estudio permitió que los pacientes mantuvieran su tratamiento con antitrombóticos, pero se excluyó a aquellos en anticoagulación con antagonistas de la vitamina K, de igual modo que a quienes tenían patología cardiovascular de relevancia clínica o antecedentes de ictus o hemorragia intracraneal. En conjunto, las principales características demográficas y clínicas basales fueron las siguientes: mediana de edad de 70 años (rango 41-91), 62% eran varones, 93% eran de raza caucásica, un 94% tenían buen estado funcional (ECOG de 0-1), la mediana de tiempo desde el diagnóstico de la enfermedad fue de 29 meses y se verificó la presencia de ganglios inflamados en el 32% de los pacientes. Las principales alteraciones citogenéticas de presencia exclusiva fueron: deleción de 17p (9%), deleción de 11q (17%), mutación de TP53 (11%), e IGHV no mutado (63%).
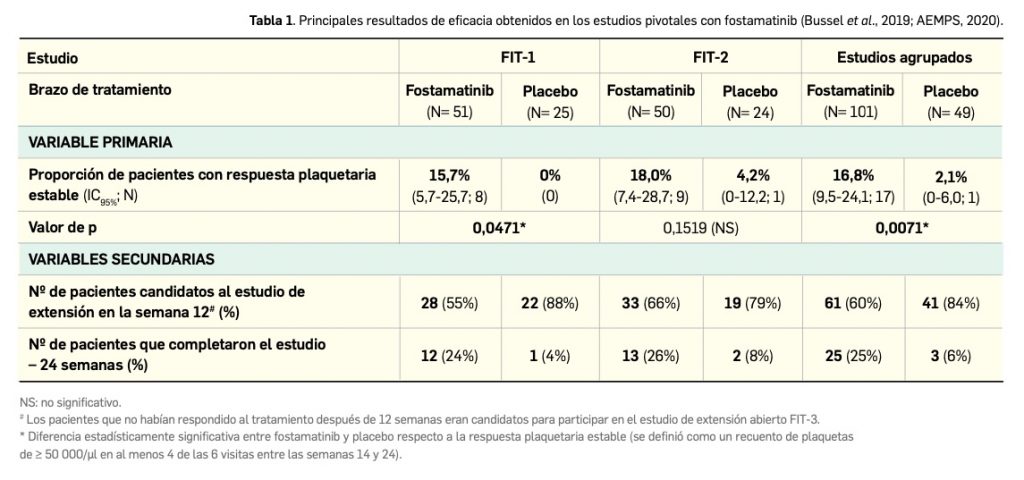
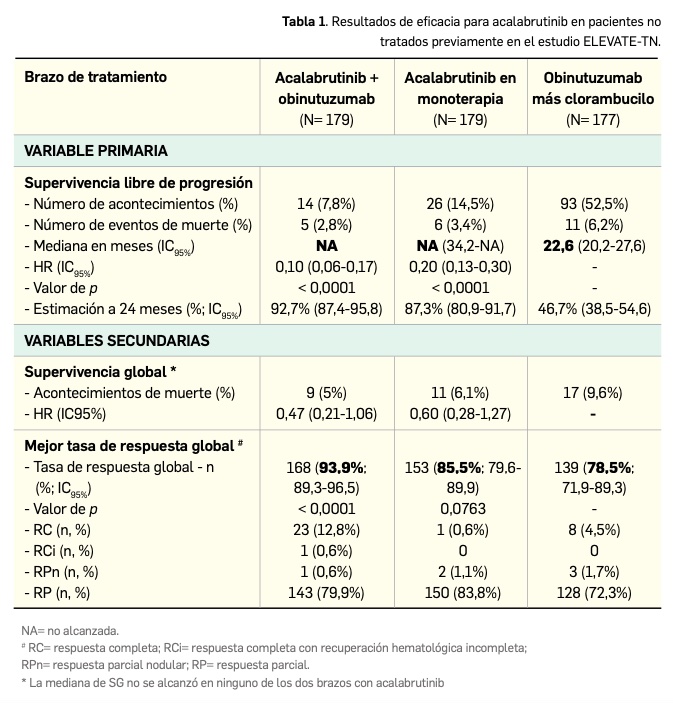
Los principales resultados de eficacia, tras una mediana de seguimiento de 28,3 meses, se presentan en la Tabla 1, habiéndose considerado como variable principal la supervivencia libre de progresión (SLP) tras revisión por comité independiente –según criterios del Grupo de Trabajo Internacional para la Leucemia Linfocítica Crónica (IWCLL)– en el brazo de acalabrutinib+obinutuzumab frente al brazo control de obinutuzumab+clorambucilo (Sharman et al., 2020). Las curvas de Kaplan‑Meier para la SLP se separan notablemente tras 8-10 meses desde la aleatorización. De modo interesante, los resultados para acalabrutinib fueron consistentes en todos los subgrupos de pacientes, incluidos aquellos con características de alto riesgo citogenético (deleción de 17p, deleción de 11q, mutación de TP53 o IGHV no mutado): la razón de riesgos o hazard ratio a favor de acalabrutinib con o sin obinutuzumab frente a obinutuzumab más clorambucilo fue de 0,08 (IC95% 0,04-0,15) y 0,13 (IC95% 0,08-0,21), respectivamente.
Por otra parte, el estudio ASCEND fue el estudio pivotal de fase 3 que permitió contrastar la eficacia y seguridad clínicas de acalabrutinib en pacientes con LLC que habían recibido al menos un tratamiento previo (excepto inhibidores de la BCL‑2 ni inhibidores del receptor de los linfocitos B) y se encontraban en refractariedad o recaída de la patología. Fue un ensayo abierto, simple ciego, multicéntrico y multinacional (159 centros en 26 países), que aleatorizó (1;1) a 310 pacientes adultos a recibir acalabrutinib en monoterapia (100 mg/12 h hasta progresión de la enfermedad o toxicidad inaceptable) o bien un control a elección del investigador6, con una combinación de idelalisib más rituximab o bendamustina más rituximab. Tras la progresión confirmada, 35 pacientes aleatorizados al brazo control cruzaron a acalabrutinib.
Como en el ensayo en pacientes naïve, se permitió el tratamiento con antitrombóticos pero se excluyó a los pacientes anticoagulados con agentes anti-vitamina K. Las características demográficas y clínicas basales más relevantes para el conjunto de pacientes fueron las siguientes: mediana de edad de 68 años (rango 32-90), 68% eran varones, 92% eran de raza caucásica, un 87% tenían buen estado funcional (ECOG de 0-1), la mediana de tiempo desde el diagnóstico de la enfermedad fue de 82 meses y se verificó presencia de ganglios inflamados en el 49% de los pacientes. Las principales alteraciones citogenéticas de presencia exclusiva fueron: deleción de 17p (16%), deleción de 11q (27%), mutación de TP53 (23%), e IGHV no mutado (78%); tenían cariotipo complejo (≥ 3 anomalías) el 31% de los pacientes. El número de tratamientos previos para la LLC fue de 1 para el 48% de los pacientes, de 2 para el 28% de los pacientes, de 3 para el 13% de los pacientes y de ≥ 4 en el 11%.
Los principales resultados de eficacia, tras una mediana de seguimiento de 16,1 meses, se presentan en la Tabla 2 (Ghia et al., 2020); de nuevo, la variable principal de eficacia fue la SLP tras revisión por comité independiente según criterios del IWCLL. Las curvas de Kaplan‑Meier para la SLP se separan notablemente tras unos 9 meses desde la aleatorización. La eficacia del nuevo fármaco se mostró consistente en todos los subgrupos, siendo independiente de la presencia de factores citogenéticos de alto riesgo; así, para estos pacientes, el hazard ratio para SLP fue de 0,25 (IC95% 0,16-0,38).
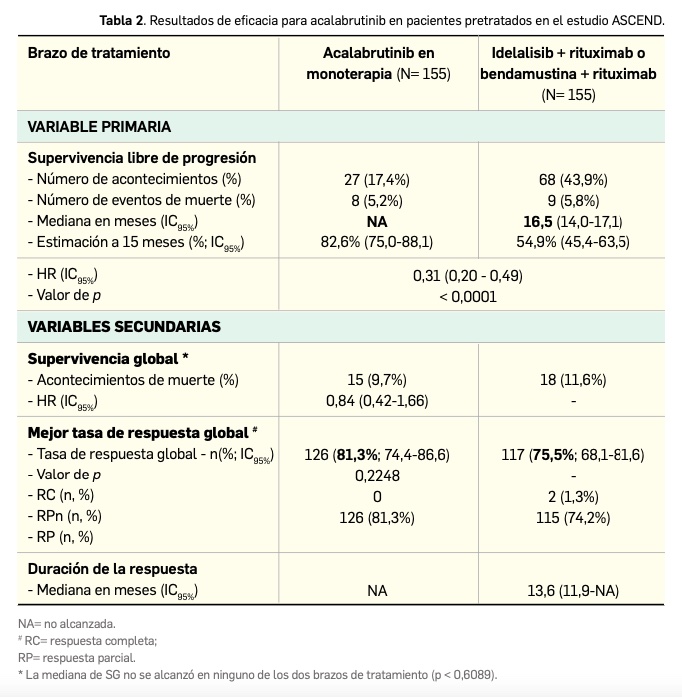
Una posterior evaluación por el investigador con un mayor periodo de seguimiento (mediana de 22,1 meses para acalabrutinib y 21,9 meses para los tratamientos usados como comparadores) demostró que la eficacia del fármaco se mantiene en el tiempo: la mediana de SLP no se alcanzó con acalabrutinib y fue de 16,8 meses para el grupo control, siendo el hazard ratio en la comparación entre grupos de 0,27 (IC95% 0,18-0,40), lo que supone una reducción del 73% en el riesgo de muerte o progresión para los pacientes asignados al brazo experimental.
Por último, los datos clínicos que han permitido caracterizar la seguridad del fármaco derivan fundamentalmente de 1.040 pacientes que lo recibieron en monoterapia durante su desarrollo clínico, con una mediana de duración del tratamiento de aproximadamente 26 meses. Las reacciones adversas de cualquier grado descritas con mayor frecuencia tras el uso de acalabrutinib fueron: infección (67%), dolor de cabeza (38%), diarrea (37%), hematomas (34%), dolor musculoesquelético (33%), náuseas (22%), fatiga (21%), tos (21%) y erupción cutánea (20%). Entre las más graves (grado ≥ 3) sobresalen: infección7 (18%), leucopenia (14%), neutropenia (14%) y anemia (8%). La tasa de interrupción del tratamiento por motivos de seguridad fue del 9,3%, destacando como causas la incidencia de neumonía, trombocitopenia y diarrea; otro 4,2% de los pacientes requirió ajustes posológicos por reacciones adversas tales como reactivación de la hepatitis B, sepsis y diarrea. No se observó una mayor tasa de mortalidad en los pacientes tratados con acalabrutinib en comparación con otras opciones antineoplásicas frente a LLC.
Cabe destacar que la combinación de acalabrutinib con otros agentes antineoplásicos fue ligeramente peor tolerada, con una frecuencia de reacciones adversas superior, si bien el tipo de eventos fue similar (infección –74%, dolor musculoesquelético –45%, diarrea –44%, cefalea –43%, leucopenia –32%, neutropenia –32%, tos –31%, fatiga –31%, etc.); la tasa de interrupción del tratamiento fue del 10,8% y la de reducción de dosis del 6,7%. En general, no se observaron diferencias clínicamente relevantes en cuanto a la seguridad o la eficacia entre los pacientes de ≥ 65 años y los de menos.
Aspectos innovadores
Acalabrutinib es un nuevo antineoplásico que actúa como un inhibidor de segunda generación, potente y altamente selectivo, de la tirosina cinasa de Bruton (BTK). Dado que la BTK participa en la señalización bioquímica del receptor de antígenos (BCR) y del receptor de citocinas de las células B, favoreciendo la supervivencia y proliferación de linfocitos B tumorales, la inhibición covalente –irreversible– que inducen acalabrutinib y su metabolito activo ACP-5862 mediante su actuación sobre el centro activo de la enzima se traduce en una inhibición de la adhesión y migración dependiente de integrinas de dichos linfocitos, conducente a la reducción tumoral. El medicamento ha sido autorizado para el tratamiento por vía oral (100 mg/2 veces al día) en monoterapia o en combinación con obinutuzumab de pacientes adultos con leucemia linfocítica crónica (LLC) no tratados previamente, y también para su uso en monoterapia en pacientes adultos con LLC que han recibido al menos un tratamiento previo.
La eficacia y seguridad clínicas del fármaco han sido adecuadamente contrastadas en dos estudios pivotales aleatorizados y multicéntricos, de fase 3 y diseño similar (abierto, simple ciego y de grupos paralelos).
Uno de ellos (ELEVATE-IN) incluyó a pacientes adultos (N= 535 pacientes de ≥ 65 años o < 65 años con comorbilidades) con LLC naïve o no tratados previamente, quienes recibieron acalabrutinib con obinutuzumab, monoterapia de acalabrutinib, o bien una combinación de obinutuzumab y clorambucilo como grupo control (comparador activo). Tras una mediana de seguimiento de 28 meses, el tratamiento con el nuevo fármaco mostró una superioridad notable en términos de SLP en comparación con el control: sin haberse alcanzado la mediana en los brazos del fármaco (vs. 22,6 meses en el grupo control), la administración de acalabrutinib con o sin obinutuzumab redujo en un 90% y un 80% el riesgo de progresión o muerte por la enfermedad, respectivamente, alcanzándose estimaciones de SLP a 24 meses del 93% y del 87%. Esa eficacia fue consistente, e incluso mayor, en pacientes con factores citogenéticos de alto riesgo, como deleción de 17p, deleción de 11q, mutación de TP53 o IGHV no mutado, en quienes redujo el riesgo de progresión o muerte en un 87-92%. El beneficio con los regímenes a base de acalabrutinib sobre obinutuzumab+corambucilo se ve robustecido por la notable mejora en supervivencia global (reducción del riesgo de muerte del 40-53%) y la diferencia en las tasas de respuesta objetiva global (86-94% vs. 79%).
El otro estudio (ASCEND) incluyó a pacientes (N= 310) con LLC refractaria o en recaída tras al menos un tratamiento previo; en torno a la mitad había recibido una sola línea de tratamiento y casi el 30% de los pacientes habían recibido 2. Con una mediana de seguimiento de 16 meses, el tratamiento con acalabrutinib en monoterapia se reveló significativamente más eficaz que un régimen antineoplásico activo –a elección del investigador8– usado como control (idelalisib + rituximab o bendamustina + rituximab). Sin alcanzarse la mediana de SLP en el brazo con el nuevo fármaco (vs. 16,5 meses en el grupo control), se estimó una reducción estadísticamente significativa del 69% en el riesgo de muerte o progresión de la enfermedad. De nuevo, la eficacia de acalabrutinib se mostró consistente en todos los subgrupos, incluyendo los pacientes con factores citogenéticos de alto riesgo, en quienes la reducción del riesgo de progresión o muerte fue del 75%. La tendencia favorable en SG y en tasa de respuesta refrendan la utilidad clínica del nuevo fármaco, que se mostró duradera: tras 22 meses no se alcanzó la mediana de SLP para acalabrutinib y la magnitud de la reducción del riesgo de progresión y muerte fue similar.
En términos de seguridad, acalabrutinib tiene un perfil toxicológico notable, con frecuentes y relevantes eventos adversos tanto en pacientes naïve como en pacientes refractarios/en racaída, que, grosso modo, está en línea con lo ya conocido para el otro inhibidor de BTK autorizado (ibrutinib). La incidencia de eventos adversos emergentes durante el tratamiento de intensidad de grado 3-4 fue del 50-70% (según régimen) y, si bien esa frecuencia parece similar a la descrita para otros tratamientos en LLC, supone tasas de discontinuación del tratamiento del 10-11% y de reducción de dosis del 4-7%. Las reacciones adversas más frecuentes (> 20%) fueron infección, dolor de cabeza, diarrea, hematomas, dolor musculoesquelético, náuseas, fatiga, tos y erupción cutánea, mientras que, entre las más graves, sobresalen las infecciones y las citopenias. Tampoco se puede obviar el riesgo aumentado de segundas neoplasias primarias, como sucede con otros antineoplásicos de “molécula pequeña”. No se observaron diferencias relevantes de seguridad en pacientes refractarios/recurrentes (en comparación con los no pretratados), en pacientes de ≥ 65 años o en aquellos con perfil citogenético de peor pronóstico.
Es preciso recordar que, en pacientes naïve, el estándar de tratamiento de la LLC para pacientes jóvenes o con buen estado de salud general es la quimioinmunoterapia basada en fludarabina, mientras que para pacientes mayores o con comorbilidades –como los incluidos en el estudio pivotal específico–, el régimen de elección está menos definido: se acepta el uso de obinutuzumab+clorambucilo, recomendándose ibrutinib si hay mutación de TP53. En el contexto de enfermedad refractaria o en recaída, se prefieren por su eficacia y menor toxicidad –frente a quimioterapia– las terapias dirigidas a marcadores de células B, como venetoclax, ibrutinib e idelalisib, siendo también una alternativa la combinación de bendamustina y rituximab (EMA, 2020).
Lo anterior determina que probablemente la selección de los comparadores en los estudios pivotales de acalabrutinib podría haber sido mejorable, aunque ha sido considerada como aceptable por la EMA. No obstante, se trata de la primera vez que se comparan en estudios clínicos de LLC en recaída/refractariedad dos de las nuevas moléculas (acalabrutinib frente a idelalisib en el estudio ASCEND), con ventaja evidente en términos de eficacia y seguridad para la primera de ellas. Además, aunque no existan evidencias que avalen su eficacia en el paciente joven y/o sin comorbilidades, parece razonable que en los pacientes en primera línea se pueda utilizar en todos los casos.
En definitiva, se trata de un inhibidor de BTK de segunda generación, cuya mayor selectividad por la enzima hace que sea al menos tan eficaz como ibrutinib pero con un perfil de toxicidad mejorado; presenta, por ejemplo, un menor riesgo de eventos adversos off target a nivel cardiaco (por ejemplo, el potencial para inducir arritmias). Sin embargo, no se dispone aún de comparaciones directas o indirectas entre ambos, y habrá que esperar a un estudio ahora en marcha en pacientes con LLC en recaída/refractariedad y alto riesgo citogenético (incluyendo del17p y del11q) para confirmar sus diferencias en eficacia y seguridad. En términos de adherencia, parece que el régimen de dos administraciones diarias es menos beneficioso que la pauta única diaria de ibrutinib.
Por ahora, es probable que el uso de acalabrutinib, tanto en monoterapia como asociado a obinutuzumab, se considere una opción preferencial en primera línea para los pacientes con LLC no tratados, similar al uso de ibrutinib o venetoclax (mayoritariamente asociados a obinutuzumab). En pacientes con LLC pretratados (incluso con ibrutinib o venetoclax) la monoterapia con acalabrutinib viene a completar las posibilidades de tratamiento junto a ibrutinib o venetoclax más rituximab, aportando una respuesta clínica relevante en cuadros con escasas opciones terapéuticas, incluso cuando éstas no son funcionales. A la vista de lo demostrado para ibrutinib, es previsible que en un futuro se demuestre un beneficio clínico con acalabrutinib en otras neoplasias de células B.
Valoración

Fármacos relacionados registrados en España