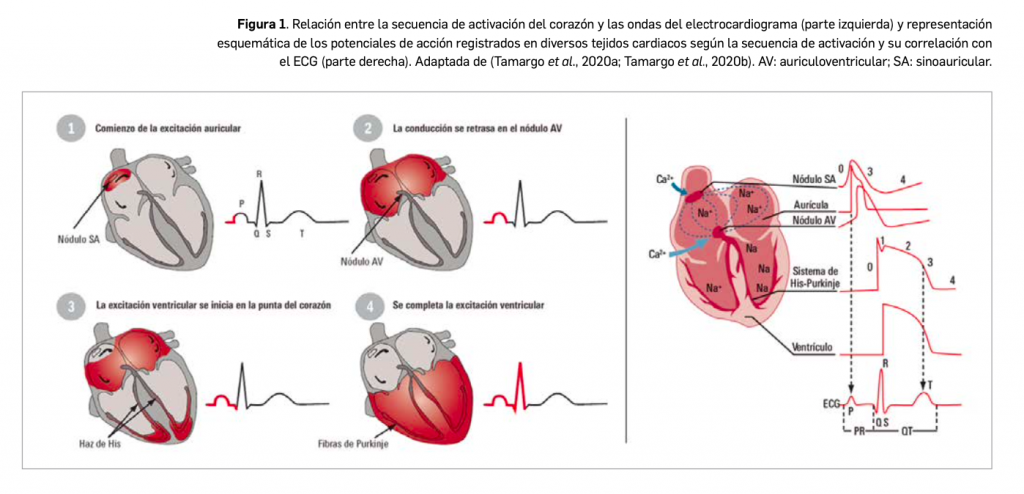
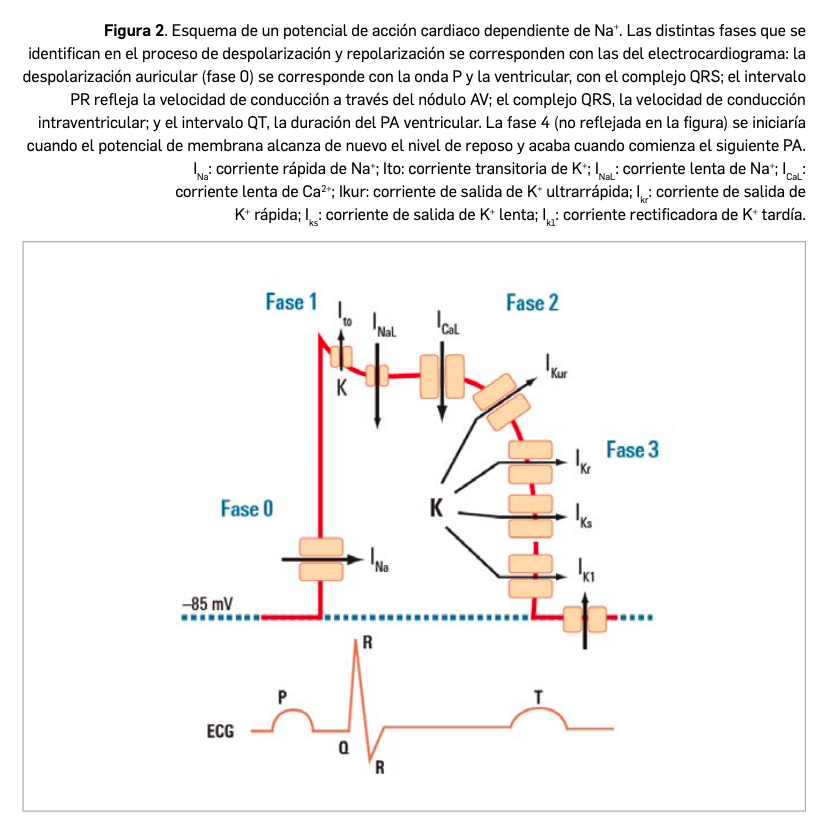
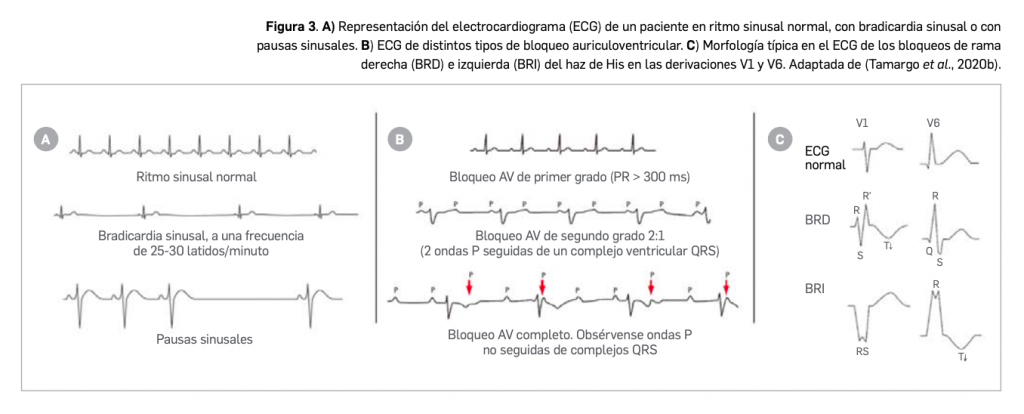
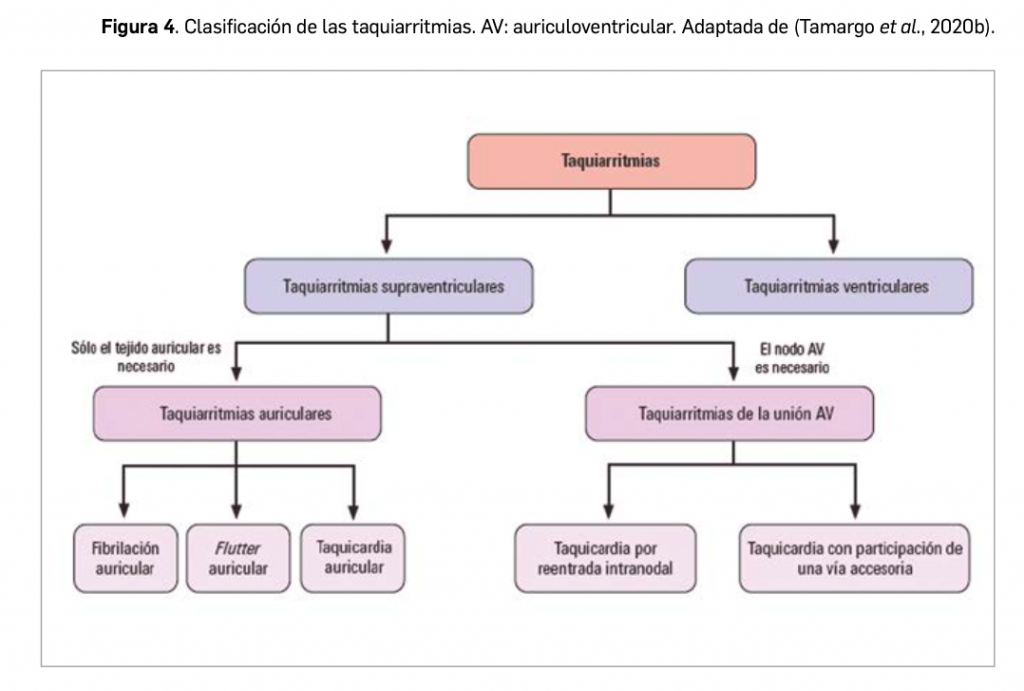
El cáncer de pulmón presenta dos tipos diferenciados: microcítico o de células pequeñas, que se observa solo en el 10%-15% de todos los tumores malignos de pulmón, y el de células no pequeñas (CPCNP), que abarca el 85%-90% restante. Éste último se divide a su vez, en función de su histología, en carcinoma de células escamosas o epidermoide -que es la forma más frecuente y se observa principalmente en hombres-, adenocarcinoma -presentado principalmente en mujeres y en nunca fumadores- y carcinoma indiferenciado de células grandes, que es el menos frecuente. Finalmente, la clasificación del estadio del cáncer se basa en las siglas T (presencia y tamaño del Tumor primario), N (presencia de metástasis en ganglios) y M (presencia de metástasis a distancia).
Se recomienda que el tratamiento del CPCNP en estadio T 2-4 y N 0-1 se base en la cirugía inicial, seguido de una quimioterapia adyuvante (QA), para eliminar las células que pudieran permanecer viables; asimismo, se puede considerar la pauta de iniciar con una quimioterapia neoadyuvante (QNA), para reducir el tamaño del tumor, continuando con cirugía. Aunque se ha visto que la pauta con QNA conduce a resultados similares respecto de supervivencia general y libre de progresión, no existen evidencias robustas sobre los beneficios; sin embargo, sí pueden hallarse diferencias respecto de los costes.
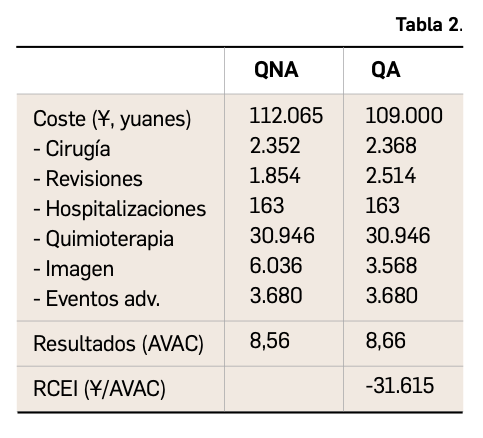
En base a lo expuesto, los autores de un reciente estudio han estimado la eficiencia de la QA versus la QNA en el tratamiento del CPCNP, bajo la perspectiva del pagador del sistema de salud. El estudio se basa en los resultados de costes sanitarios directos (cirugía, revisiones de esta, hospitalizaciones, quimioterapia, diagnósticos por imagen y manejo de eventos adversos) y beneficios (supervivencia general y años de vida ajustados a calidad, AVAC), que se implementan en un árbol de decisión. Dicho modelo analiza la QA en 4 ciclos respecto de la QNA en 2 ciclos. En ambas se asume que cada ciclo está compuesto por un régimen intravenoso de paclitaxel/carboplatino; asimismo, se asume que se presentarán eventos adversos asociados a la quimioterapia, en grados 3 o 4, al mismo tiempo que se asume la posibilidad de problemas quirúrgicos en ambos grupos. Se consideraron como puntos finales del estudio la recuperación o el fallecimiento de los pacientes. Como en algunos estudios no se mostraron diferencias significativas respecto de la supervivencia global en QA y QNA, el modelo incluye un valor de 9,1 años para ambas, manteniendo asimismo la misma calidad de vida (con valores de utilidad extraídos de la bibliografía).
Los resultados de eficiencia, estimados mediante el ratio coste-efectividad incremental (RCEI) mostraron dominancia con la estrategia de QA, al presentar mejores resultados y menores costes (Tabla 2). Los análisis de sensibilidad determinísticos mostraron que el resultado del RCEI fueron muy sensibles al valor de la supervivencia general de la QNA. Los análisis probabilísticos estimaron que, para un umbral de 35.446 ¥/AVAC, la estrategia con QA presentaría una probabilidad del 54% de ser coste-efectiva.
A la vista de los resultados, los autores concluyen que, a lo largo de la vida, la quimioterapia adyuvante posterior a la cirugía presenta un mejor valor de eficiencia que la quimioterapia neoadyuvante, previa a la cirugía, en el tratamiento inicial del cáncer de pulmón de células no pequeñas.