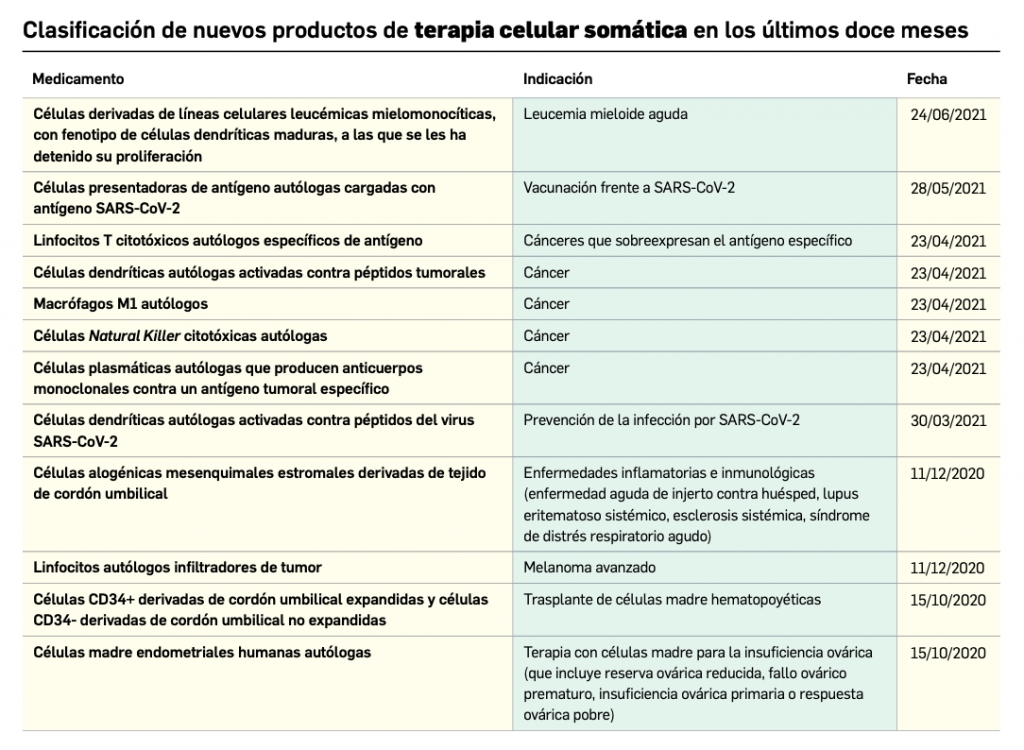
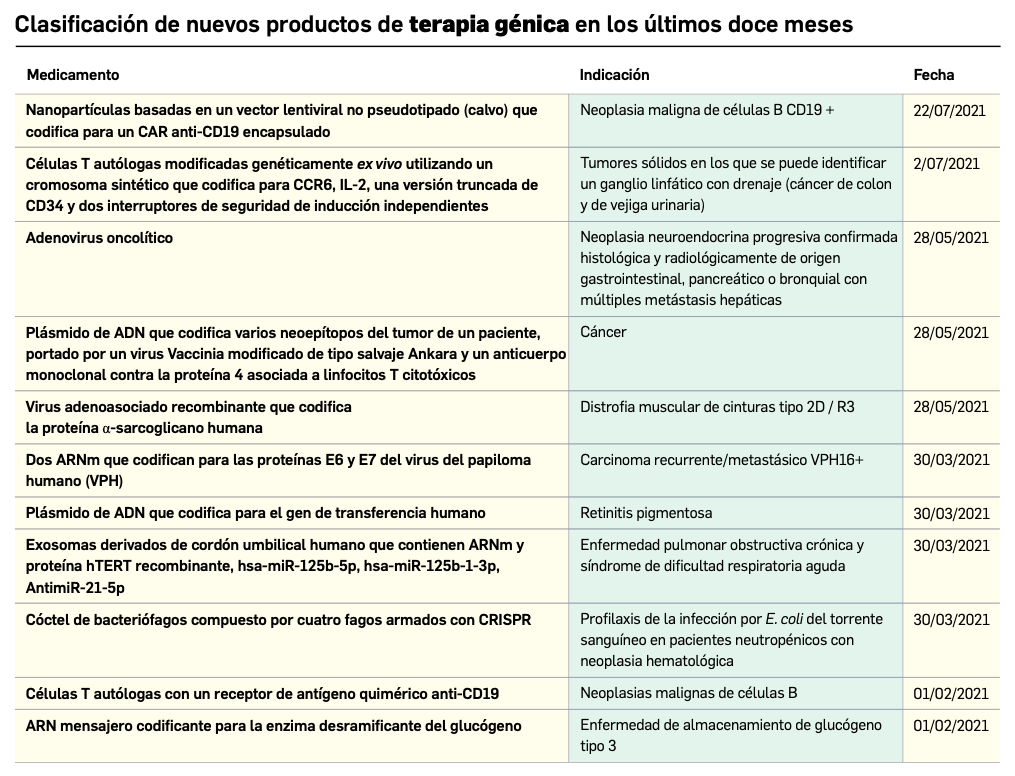
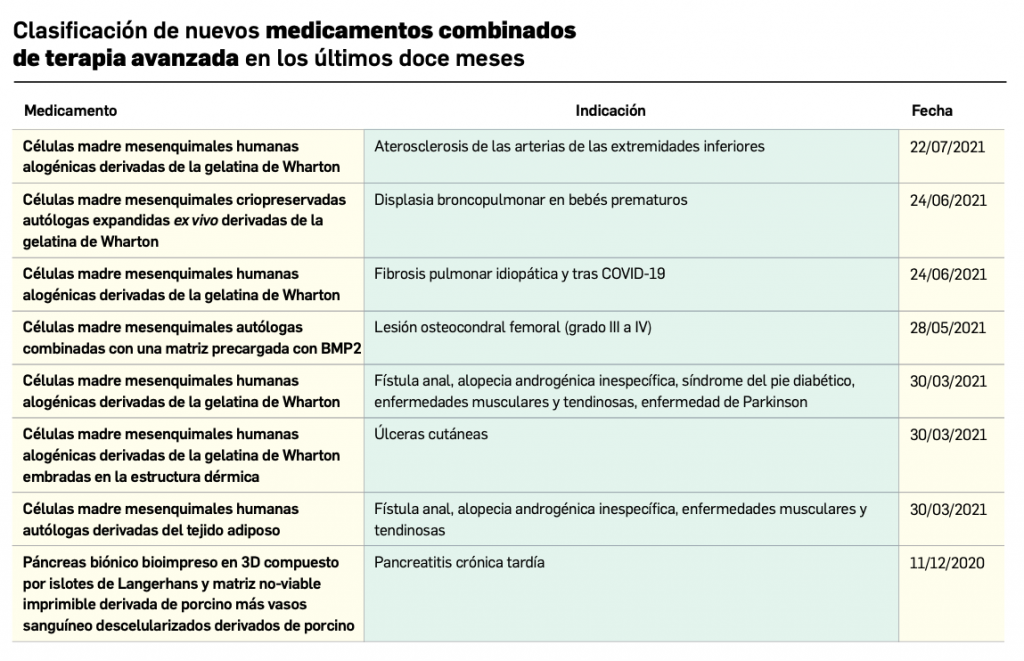
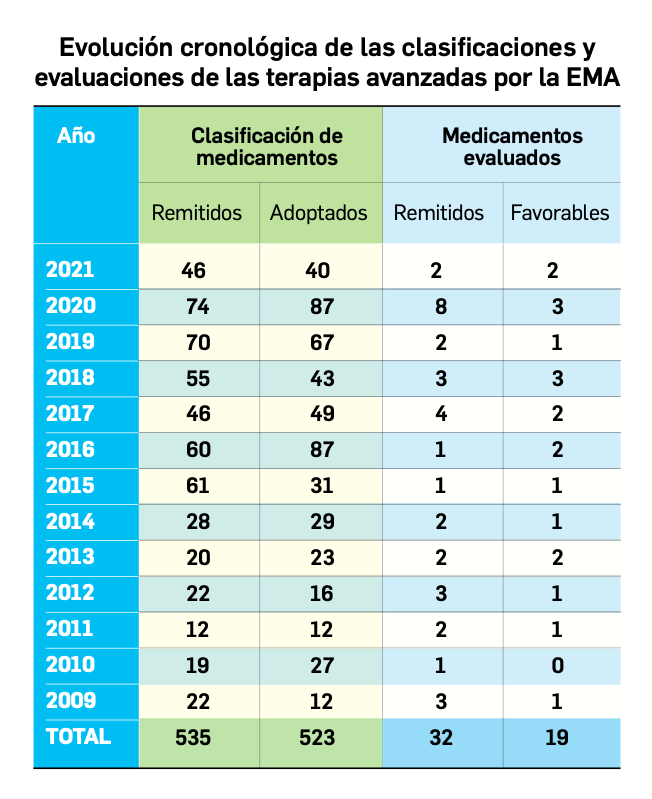
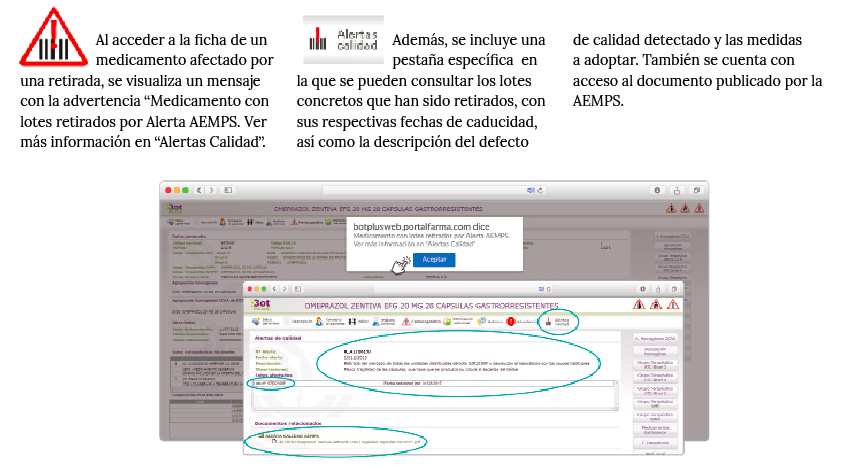
Desde que comenzó la pandemia por COVID-19 en marzo de 2020, diversos investigadores han sospechado que los pacientes que estaban vacunados contra la gripe respondían mejor a la infección por SARS-CoV-2.
Yang y colaboradores fueron los primeros en realizar un estudio con 2.005 pacientes. Hicieron una revisión retrospectiva y encontraron un posible efecto protector de la vacunación antigripal frente a las complicaciones asociadas a la infección por SARS-CoV-2.
Aunque no se ha demostrado una reactividad cruzada entre los anticuerpos inducidos por la gripe y la protección frente a SARS-CoV-2, en la literatura reciente se han propuesto varios mecanismos teóricos al respecto. La primera hipótesis se centra en la presencia de MF59: se ha observado que este adyuvante potencia una respuesta inmunitaria frente a las variantes del SARS-CoV. Alternativamente, el efecto protector potencial de la vacuna puede explicarse por su capacidad para estimular la activación de las células NK (natural killers), cuyos niveles se han encontrado considerablemente disminuidos en los casos moderados y graves de COVID-19.
Otro mecanismo propuesto se describió en un reciente estudio de casos y controles con 261 trabajadores de la salud. En él, los autores sugirieron que tanto los coronavirus como los virus de la gripe se relacionan con la enzima convertidora de angiotensina 2 (ACE-2) y los anticuerpos de tetraspanina (proteínas de membrana). Los anticuerpos ACE-2 y tetraspanina pueden inhibir tanto las infecciones por coronavirus como por el virus de la gripe A de baja patogenicidad. Los resultados de este estudio apuntaron también a un efecto protector potencial en aquellos con vacunación antigripal. Estudios adicionales indican que la vacuna contra la gripe puede reducir el riesgo de eventos cardiovasculares debido a la posible interacción con los sistemas inmunitario e inflamatorio para promover la estabilización de la placa de ateroma. También se ha postulado recientemente que los anticuerpos inducidos por la vacuna contra la gripe pueden interactuar con el receptor de bradiquinina-2, lo que lleva a un efecto antiinflamatorio secundario al aumento de óxido nítrico.
Con estos antecedentes, varios investigadores americanos han llevado a cabo un estudio a partir de los registros de más de 70.000 pacientes de distintos países, incluidos Estados Unidos, Reino Unido, Alemania, Italia, Israel y Singapur. Los investigadores examinaron en primer lugar los datos de más de 70 millones de pacientes, a partir de los cuales identificaron dos cohortes de 37.377 pacientes. El primer grupo había recibido la vacuna de la gripe entre dos semanas y seis meses antes de ser diagnosticado de COVID-19, mientras que los del segundo grupo no fueron vacunados. Ambos grupos se emparejaron por factores como grave, como edad, sexo, etnia, tabaquismo y comorbilidades como diabetes, obesidad y EPOC, para afinar la comparativa. La vacuna de la gripe en pacientes con COVID-19 se relacionó con una reducción del riesgo de ictus, sepsis y trombosis venosa profunda. Los pacientes que habían sido vacunados visitaron menos las urgencias y presentaron una menor probabilidad de acabar ingresados en la UCI. Sin embargo, no se halló una reducción de la mortalidad. Para determinar de forma más firme la posible asociación entre la vacuna de la gripe y la protección frente a la infección por SARS-CoV-2, los científicos señalan que es necesario llevar a cabo más ensayos clínicos prospectivos.