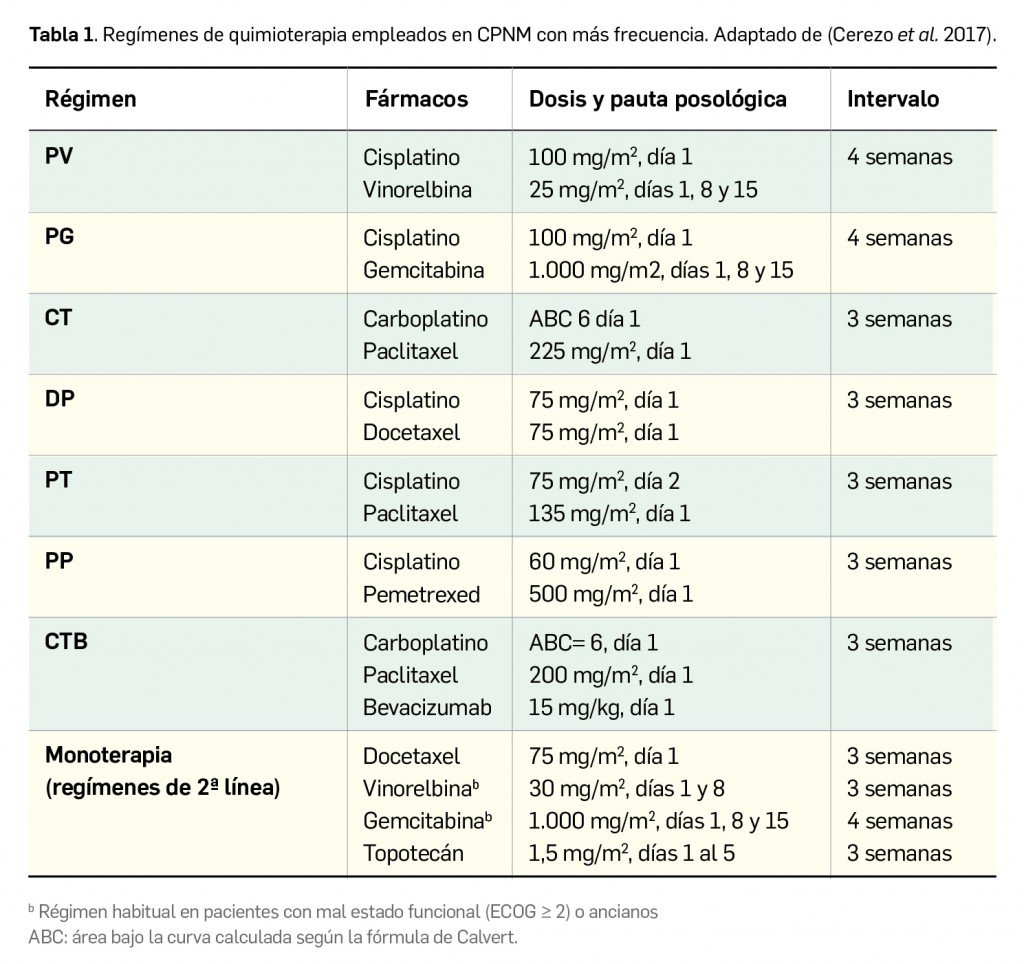
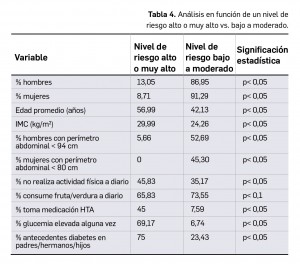
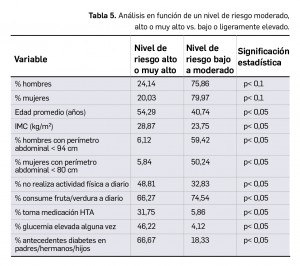
Los resultados recientemente divulgados del estudio SURPASS-2 muestran que tirzepatida es capaz, en una pauta de una dosis semanal por vía subcutánea, de reducir la hemoglobina glicosilada tras 40 semanas de tratamiento en mayor medida que semaglutida, con una diferencia significativa de -0,15 a -0,45 puntos porcentuales según la dosis. Además, proporciona una reducción notable del peso (de -1,9 a -5,5 kg de diferencia) con un perfil de seguridad similar al del resto de análogos de GLP-1, caracterizado fundamentalmente por eventos adversos gastrointestinales leves-moderados, sin problemas de hipoglucemia clínicamente significativa.
Tirzepatida es un fármaco en investigación clínica frente a la diabetes mellitus tipo 2 (DM2) con un mecanismo de acción dual: actúa como agonista del receptor del polipéptido insulinotrópico dependiente de glucosa (GIP) y como un agonista del receptor del péptido-1 similar a glucagón (GLP-1), que integra en una única molécula los efectos biológicos de ambas incretinas. Su eficacia y seguridad en una pauta de una vez por semana han sido contrastadas en un amplio estudio de fase 3 (SURPASS-2), abierto, de 40 semanas de duración, controlado con semaglutida –otro análogo de GLP-1 comercializado en España por primera vez en 2019 que ha mostrado una eficacia hipoglucemiante parcialmente superior a otros fármacos de su grupo (por ejemplo, exenatida, liraglutida o dulaglutida). El estudio aleatorizó a un total de 1.879 pacientes (1:1:1:1) a recibir tirzepatida a las dosis semanales de 5, 10 o 15 mg, o semaglutida a la dosis de 1 mg/semana, ambos en combinación con metformina; consideró como variable primaria de eficacia el cambio a la semana 40 del nivel de hemoglobina glicosilada (HbA1c). Cabe destacar, entre las características basales de la población incluida (equilibradas entre los brazos de tratamiento), que el nivel medio de HbA1c fue de 8,28%, la edad media de 56,6 años, y el peso medio de 93,7 kg.
Los resultados revelan que tirzepatida es no-inferior y superior a semaglutida en la reducción de la HbA1c en todas sus dosis: el cambio medio desde el nivel basal fue de -2,01, -2,24 y -2,30 puntos porcentuales, respectivamente, para los tres niveles de dosis ascendentes del fármaco, frente a -1,86 puntos para semaglutida. Así, la diferencia estimada con el comparador activo fue de -0,15 puntos porcentuales para la dosis de 5 mg/semana (IC95% -0,28 a -0,03; p= 0,02), -0,39 para la de 10 mg/semana (IC95% -0,51 a -0,26; p< 0,001) y de -0,45 puntos para la de 15 mg/semana (IC95% -0,57 a -0,32; p< 0,001); uno de cada dos pacientes tratados con tirzepatida 15 mg/semana alcanzó niveles de HbA1c similar al de personas no diabéticas, de < 5,7% (vs. el 22% de pacientes con semaglutida). Además, la reducción media en el peso corporal también fue significativamente mayor con tirzepatida (-1,9, -3,6 y -5,5 kg, respectivamente, de diferencia con semaglutida; p< 0,001 en todas las comparaciones). En relación a la seguridad, el perfil parece consistente con el de otros análogos de GLP-1: los eventos adversos más comunes con ambos fármacos fueron gastrointestinales y en su mayoría leves-moderados, destacando por su frecuencia con tirzepatida las náuseas (17-22%), la diarrea (12-16%), y los vómitos (6-10%); los eventos adversos graves fueron solo ligeramente más incidentes que con semaglutida (5-7% vs. 3%). El riesgo de hipoglucemia con el nuevo fármaco parece bajo (0,6-1,7% vs. 0,4% con semaglutida).
En definitiva, la diabetes, por su elevada prevalencia a nivel mundial, sigue ocupando los esfuerzos de muchos investigadores. Los hallazgos aquí resumidos, consistentes con los previamente divulgados para tirzepatida, son una nueva muestra de ello y pueden contribuir a ampliar próximamente las opciones de farmacoterapia disponibles. Es preciso subrayar, no obstante, que los agonistas del receptor de GLP-1 ocupan, según las principales guías de práctica clínica, su lugar en las segundas o terceras líneas de tratamiento de la DM2, siempre en combinación con metformina en terapia doble (si hay contraindicación o tolerancia a sulfonilureas) o triple (también con sulfonilureas).