Resumen
Siponimod es un modulador del receptor de la esfingosina 1-fosfato (S1P) que se une selectivamente a los receptores S1P1 y S1P5. Al actuar como un antagonista funcional en los receptores S1P1 de los linfocitos, que se insensibilizan de forma dosis-dependiente al efecto de la S1P, el fármaco previene la salida de dichas células desde los órganos linfoides y, en consecuencia, provoca una redistribución linfocitaria: quedan “secuestrados” y se reduce su infiltración patológica al sistema nervioso central, limitando la inflamación y lesiones en el tejido nervioso; además, siponimod reduce el recuento de linfocitos periféricos en un 20-30% respecto a niveles basales. En base a ello, el medicamento ha sido autorizado para el tratamiento por vía oral de pacientes adultos con esclerosis múltiple secundaria progresiva (EMSP) con enfermedad activa definida por brotes o por características de imagen típicas de actividad inflamatoria.
Los datos clínicos que han sustentado su autorización derivan de un único ensayo pivotal multicéntrico de fase 3 con una fase principal doble ciego y controlada, en que el tratamiento diario con siponimod demostró superioridad clínica sobre placebo en pacientes con EMSP (N= 1.651). En esa comparativa, se observó un beneficio relevante con el uso del fármaco en la población global de pacientes, plasmado en el retraso de la progresión de la discapacidad, con una reducción relativa –estadísticamente significativa– del riesgo del 21% a los 3 meses y del 26% a los 6 meses, así como en una reducción del 55% en la tasa anualizada de brotes. La superioridad del fármaco sobre placebo, consistente en todos los subgrupos analizados, fue mayor en los pacientes con enfermedad activa al inicio, en quienes redujo el riesgo de progresión de la discapacidad en un 31% y 37% a los 3 y 6 meses, respectivamente, disminuyendo en un 46% la tasa de brotes confirmados. En todo caso, la baja proporción de pacientes que progresó tras 3 años de seguimiento, la ausencia general de significación estadística en los resultados de calidad de vida reportados por los pacientes o la inclusión de pacientes con enfermedad y progresión moderada (impide probar convincentemente la prevención de la progresión de la discapacidad) contribuyen a cuestionar la relevancia clínica de los resultados. Con respecto a la seguridad, siponimod muestra un perfil toxicológico complejo, pero similar al del otro miembro de su grupo farmacológico –fingolimod–, que se asocia con una baja tasa de discontinuaciones. La incidencia de eventos adversos es escasamente superior a placebo, pero sobresalen riesgos de seguridad relativos a la frecuencia de infecciones y a los eventos adversos cardiovasculares (bradicardia o bloqueo de la conducción cardiaca), que hacen necesario un seguimiento específico de los pacientes. Por su frecuencia destacan las alteraciones del sistema nervioso, cefalea e hipertensión, mayoritariamente leves-moderadas, mientras que por su severidad se subraya el riesgo de infecciones del tracto urinario, elevaciones de ALT y depresión.
En resumen, este nuevo fármaco modificador de la enfermedad aporta un beneficio clínico superior a placebo en pacientes con EMSP y continúa la vía terapéutica inaugurada por fingolimod, sin aportar innovación a nivel mecanístico. El IPT lo considera una alternativa a interferón-β 1b, ocrelizumab o cladribina en el tratamiento de pacientes con EMSP y actividad patológica, sin ser posible establecer la superioridad de uno sobre otro; aunque la evidencia en el caso de siponimod es más robusta que para ocrelizumab o cladribina, no se puede concluir sobre su beneficio en todo el espectro de la enfermedad con EMSP. Tampoco aporta ningún otro aspecto innovador reseñable (vía de administración novedosa en la indicación o mejor perfil toxicológico).
Aspecto pisiopatológicos
La esclerosis múltiple (EM) es una enfermedad autoinmune inflamatoria y crónica del sistema nervioso central (SNC). Se caracteriza por la presencia de múltiples placas diseminadas de desmielinización focal –se la denomina también esclerosis en placas o esclerosis diseminada– distribuidas a lo largo del cerebro y la médula espinal, dando lugar a múltiples y variados síntomas y signos de disfunción del SNC (Fernández del Pozo et al., 2018).
Esta enfermedad afecta a más de 2,5 millones de personas a nivel mundial, especialmente a adultos jóvenes, y se la considera la causa de invalidez de tipo no traumático más frecuente en esta población. La edad típica de debut clínico de la enfermedad es entre los 16 y los 50 años de edad (media de 30 años), con un máximo entre los 20 y los 40, siendo muy rara su aparición antes de los 10 años o después de los 70. Es 2 veces más común en las mujeres, en quienes suele comenzar más temprano, y su prevalencia es mayor en los países del norte de Europa, siendo menos frecuente en el área mediterránea. En España, la prevalencia se sitúa en los 100-125 casos por cada 100.000 habitantes.
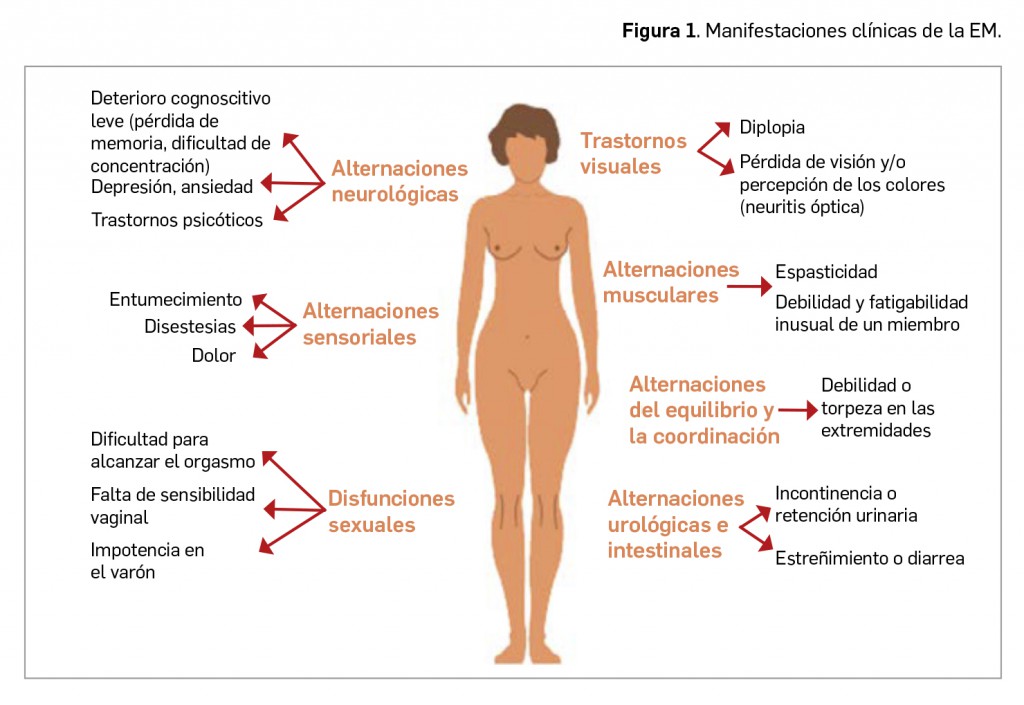
El curso de la enfermedad es variable. Se caracteriza mayoritariamente por la presencia de periodos de remisión y recaídas, con algunos efectos acumulativos, y, aunque la discapacidad neurológica puede aparecer desde el inicio de la enfermedad, lo más frecuente es que comience a manifestarse tras varios años de evolución. O sea, se trata de una enfermedad con un carácter lentamente progresivo, con remisiones y exacerbaciones recurrentes. Las manifestaciones clínicas son muy variadas, dependiendo de los nervios afectados. Pueden observarse alteraciones musculares y de la coordinación, sensoriales, alteraciones neurológicas, trastornos visuales o disfunciones sexuales.
Entre los trastornos musculares sobresale la debilidad muscular, que puede intensificarse con el calor, la fiebre o el esfuerzo. Esta debilidad se traduce también en fatiga, que se añade a la causada por los problemas de movilidad y que, si afecta a la respiración, deriva en una situación de discapacidad muy importante. Los músculos también pueden estar tensos o rígidos (espasticidad). Si afecta a nervios craneales, puede provocar parálisis facial. Además, la afectación del cerebelo puede ocasionar trastornos en el habla, temblor y dificultad de coordinación que, junto con los trastornos del equilibrio y la debilidad muscular, la espasticidad, la fatiga y las alteraciones sensoriales, contribuyen a uno de los signos más característicos de la EM: los trastornos de la marcha; éstos son muy frecuentes (aparecen en casi el 50% de los pacientes incluso en fases tempranas) y de los más limitantes para la actividad diaria de los pacientes.
Por su parte, las alteraciones sensoriales son muy frecuentes y suelen iniciarse distalmente, con hormigueo o entumecimiento en la punta de los dedos –parestesias– y posible aparición de dolor (disestesias). La afectación de algunos nervios craneales puede dar lugar a cefaleas (neuralgia del trigémino) o alteraciones del sentido del gusto y la propiocepción. La EM puede también ocasionar trastornos visuales: la inflamación del nervio óptico (neuritis óptica) da lugar a dolor ocular, que empeora con el movimiento del globo, pérdida de visión y/o percepción del color, y la parálisis de los músculos oculares puede causar diplopía. Adicionalmente, en hasta dos tercios de los casos pueden observarse trastornos de la función vesical (incontinencia en los primeros estadios que puede derivar en retención urinaria) y la función intestinal (estreñimiento, diarrea). La función sexual puede verse alterada por problemas de erección y lubricación, disminución de la excitación y pérdida de sensibilidad. Se observan, en algunos casos, alteraciones neuropsicológicas, ya que la afección del SNC puede conducir a un procesamiento más lento de la información, fallos de atención, problemas de memoria y alteración de las funciones ejecutivas.
Este amplio abanico de manifestaciones posibles de la EM (Figura 1) podrá alterar el estado emocional de la persona, aumentando en estos pacientes la incidencia de depresión y ansiedad, que empeoran algunos de los síntomas anteriores.
Con una etiología no bien conocida, el desarrollo de la EM se atribuye, como en otras patologías autoinmunes, a la exposición a factores de riesgo ambientales de individuos genéticamente predispuestos. En este caso, entre los factores genéticos de riesgo se han identificado el haplotipo HLADRB1 y algunos genes que codifican para receptores de la célula T. Entre los factores ambientales se han descrito: la infección previa por algunos virus como el de Epstein-Barr, el tabaco, las toxinas ambientales y los niveles bajos de vitamina D; las hormonas sexuales afectan también a la sintomatología y evolución de la EM, observándose un efecto protector de los estrógenos y progestágenos (la EM mejora en el embarazo) y con niveles altos de testosterona. Sea como fuere, en la patogenia de la enfermedad se consideran 2 fases: a) en primer lugar, un proceso inflamatorio autoinmune que caracteriza los años iniciales y se manifiesta por brotes, y en el que se constatan por resonancia magnética nuclear (RMN) lesiones desmielinizantes que afectan tanto a la sustancia blanca como a la gris; y b) en segundo lugar, un proceso degenerativo, consecuencia del daño neuronal irreversible, que, si bien existe desde las primeras etapas, constituirá el sustrato de la fase progresiva tardía. El estudio histopatológico de las placas muestra lesiones formadas por infiltrados de células, con desmielinización y gliosis (proliferación de astrocitos y pérdida neuronal, que da lugar a una cicatriz glial).
Se ha propuesto que un antígeno, todavía desconocido, pero probablemente de origen externo (posiblemente herpesvirus o retrovirus humanos, como el virus inotrópico de células T humanas o HTLV-1, que causa la paraparesia espástica tropical, o virus implicados en infecciones comunes, como el sarampión), desencadenaría, por mimetismo molecular con la fracción proteica de la mielina, la reacción autoinmune. Ese antígeno se presentaría sobre la superficie de los macrófagos en combinación con moléculas del MHC de clase II. La resultante estimulación de los linfocitos T cooperadores Th1 provocaría la expresión de LFA-1 y de VLA-4, facilitando la unión de dichos linfocitos T a moléculas de adhesión (como la ICAM-1 y la VCAM-1) sobre las células endoteliales de los vasos sanguíneos, facilitando su migración a través del endotelio y su penetración en el SNC.
Para la posterior destrucción de la mielina se han planteado 3 mecanismos complementarios: a) liberación de TNFα por los linfocitos Th1; b) liberación de TNFα y de radicales libres de oxígeno, óxido nítrico y proteasas por los macrófagos activados; y c) activación de la cascada del complemento mediante anticuerpos. Sea como fuere, la principal consecuencia de la destrucción de las vainas mielínicas es la alteración de la conducción de los impulsos nerviosos transmitidos por las fibras desmielinizadas. La velocidad de transmisión se hace más lenta y los estímulos no se transmiten correctamente o incluso no lo hacen en modo alguno. El grado de anormalidad de la conducción, que determina la sintomatología de la EM, puede variar dependiendo de circunstancias como la temperatura corporal (la mayoría de los pacientes experimentan un empeoramiento de sus síntomas al aumentar la temperatura), el ejercicio o la composición iónica del espacio extracelular.
La enfermedad se asocia a un primer episodio con síntomas neurológicos indicativos de desmielinización (síndrome desmielinizante aislado), que persiste durante al menos 24 horas. Esta situación no implica un diagnóstico de EM y, a partir de ella, puede desarrollarse o no EM. Dependiendo de su comportamiento clínico, la enfermedad puede clasificarse en las cuatro formas clínicas que se recogen a continuación. Esta distinción entre tipos de EM es importante, dado que no responden igual a los tratamientos disponibles y, de hecho, la mayoría de ellos carecen de efecto sobre la forma progresiva primaria.
- EM remitente-recurrente (EMRR). Afecta al 85-90% de los pacientes con EM y se caracteriza por recaídas y remisiones, que pueden ser parciales o completas. Tiene un inicio brusco, con la máxima expresión de los síntomas en unas horas o pocos días, disminuyendo de intensidad hasta remitir prácticamente de forma completa durante un largo periodo antes de un nuevo brote.
- EM secundaria progresiva (EMSP). Comienza de la misma forma que la EM remitente-recurrente, pero en algún momento se produce un deterioro continuo, sin relación con los ataques agudos o subagudos. El 50% de los pacientes con EM remitente-recurrente terminará por sufrir la forma progresiva secundaria a los 15-20 años, por lo que parece que esta forma podría ser la fase tardía de la EM remitente-recurrente (no existen criterios claros que marquen el paso de una a otra). La EMSP se diagnostica retrospectivamente por una historia clínica de empeoramiento gradual tras un curso inicial de EM recurrente1, cursando con o sin exacerbaciones agudas durante la fase progresiva; resulta difícil determinar el momento en que predomina la progresión independiente de las recaídas. En relación con la actividad de la enfermedad, las características distintivas de la actividad inflamatoria en la EMSP pueden estar relacionadas con los brotes o con la imagen, es decir, lesiones T1 realzadas con gadolinio o lesiones T2 activas (nuevas o que han crecido).
- EM primaria progresiva (EMPP). Como su nombre indica, no se manifiestan recaídas bruscas ni remisiones, sino un deterioro neurológico continuo y progresivo desde el inicio. Esta forma clínica se manifiesta en aproximadamente el 10-15% de los pacientes diagnosticados de EM.
- EM progresiva-recurrente. Representa un 5% de los casos de EM, que presentan una progresión lenta, como en la progresiva primaria, pero con brotes agudos más graves.
Aunque las remisiones asintomáticas pueden perdurar hasta más de 10 años, algunos pacientes tienen crisis frecuentes y rápidamente llegan a la incapacitación. En líneas generales, a los 5 años desde la aparición de los primeros síntomas, algo más del 50% de los pacientes tiene algún tipo de afectación leve, en otro 40% hay afectación moderada y en menos de un 10% es grave; se estima que en torno al 70% de los pacientes está en condiciones de trabajar habitualmente. No obstante, a los 15 años desde el inicio de los síntomas solo el 25-30% de los pacientes continua con una afectación leve y un 50% requiere ayuda para caminar. A los 20 años, solo un 35% continua en condiciones de trabajar y un 20% ha muerto como consecuencia de las complicaciones.
Se acepta que la EM produce globalmente una reducción media sobre la duración de la vida en los varones de unos 9 años y hasta de 14 años en las mujeres. La esperanza de vida media suele rondar los 25 años tras el comienzo de la enfermedad, aunque con notables variaciones interindividuales. El pronóstico depende fundamentalmente del número de ataques, siendo un signo de mal pronóstico la existencia de una elevada frecuencia de recaídas durante los primeros años de enfermedad (la frecuencia media de es de 1 ataque anual al principio). Igualmente, el tipo de ataques es relevante, pues los síntomas primarios de tipo motriz, ataxia o problemas bulbares se asocian con peores pronósticos, mientras que, si son de tipo visual, el pronóstico es más favorable.
Tratamiento
Desde el punto de vista de su terapéutica, en la actualidad tiende a hablarse de formas de EM que cursan con/sin
actividad (episodios agudos de disfunción neurológica, y/o aparición de nuevas lesiones o captación de contraste y/o progresión).
El tratamiento de la EM tiene como objetivos: reducir la gravedad y la frecuencia de las recaídas, limitar la discapacidad persistente, aliviar los síntomas y promover la reparación tisular. Para ello, el abordaje del paciente debe contemplar tanto farmacoterapia como tratamiento rehabilitador físico y neuropsicológico. Este último mejora la capacidad motora y la calidad de vida de los pacientes; supervisado por el neurólogo, se adecuará a la situación del paciente y puede incluir fisioterapia, atención psicológica, terapia ocupacional y logopedia. En cuanto a la farmacoterapia, no existe por el momento ningún tratamiento curativo de la enfermedad y se aborda desde 3 perspectivas: i) tratamiento de fase aguda, que reduzca la gravedad del brote y su repercusión posterior; ii) tratamiento modificador de la enfermedad, para el que se dispone de cada vez más fármacos, que actúan a diferentes niveles; y iii) tratamiento sintomático, para aliviar la sintomatología y mejorar la calidad de vida del paciente.
Atendiendo al carácter inflamatorio e inmunológico de la EM, el tratamiento de elección para las recaídas agudas son los corticosteroides. Reducen la intensidad y la duración de la recaída, probablemente reduciendo el edema, pero no afectan a la progresión de la discapacidad.
La terapia modificadora de la enfermedad busca reducir la frecuencia y la intensidad de los ataques y prevenir la acumulación de discapacidad. En la actualidad, se encuentran autorizados en la Unión Europea varios fármacos modificadores de la enfermedad (FAMEs), entre los que se dispone de agentes inmunomoduladores (beta-interferones, peginterferón beta-1a, acetato de glatirámero), anticuerpos monoclonales (antagonistas de la alfa-4-beta integrina [natalizumab] y anti-CD52 [alemtuzumab]), agentes inmunosupresores y citotóxicos. Entre las terapias orales se encuentran fingolimod (análogo de esfingosina), teriflunomida (inhibidor de la síntesis “de novo” de pirimidinas) y dimetilfumarato (derivado del ácido fumárico que activa la vía de transcripción del factor nuclear 2) y cladribina (análogo nucleósido de desoxiadeosina) (AEMPS, 2021). Otros tratamientos usados en el pasado para tratar la EM, como mitoxantrona o azatioprina han caído en desuso. Hasta la comercialización en 2019 de ocrelizumab (un anticuerpo monoclonal anti-CD20), los fármacos disponibles en la UE solo eran útiles en la forma remitente-recurrente y algunos en la progresiva secundaria, pero no en la progresiva primaria en que se indica dicho fármaco.
Los fármacos inmunomoduladores empleados en el tratamiento de las formas recurrentes de EM son el interferón (IFN) beta y el acetato de glatirámero. El IFN-β(1a y 1b) ha demostrado un efecto beneficioso en cuanto a reducción del número y la gravedad de las recaídas, así como una importante mejoría de las lesiones. El IFN-β 1b está indicado en pacientes ambulatorios con formas remitentes-recurrentes de EM, capaces de andar, para reducir la frecuencia y la gravedad de las recaídas. También está indicado para ralentizar la progresión de la enfermedad y reducir la frecuencia de las exacerbaciones clínicas, en pacientes con y sin crisis y en todos los niveles de incapacidad investigados (no se ha estudiado a pacientes leves ni a aquellos que no podían andar). El tratamiento de 2 años en estos casos ha demostrado un significativo incremento del tiempo hasta la progresión de la enfermedad, así como otros importantes beneficios, tales como el retraso de la necesidad de usar silla de ruedas, reducir el consumo de corticosteroides y el número de hospitalizaciones.
Por su parte, el IFN-β 1a está indicado en el tratamiento de pacientes ambulatorios con formas remitentes-recurrentes de EM, capaces de andar, en los que reduce la frecuencia (en un 30%) y la gravedad de las recidivas clínicas, así como el número de hospitalizaciones por la enfermedad. Se observa además una prolongación del intervalo sin enfermedad, pero no un efecto importante sobre la progresión de la enfermedad. El peginterferón-β 1a tiene la misma indicación y presenta una semivida más prolongada que el IFN no pegilado. Hasta hace poco, las dos formas de interferón (INF-β1b e INF-β1a) eran los únicos medicamentos disponibles para el tratamiento de las formas secundariamente progresivas con brotes.
El acetato de glatirámero es una mezcla de péptidos sintéticos formados por copolímeros de ácido L-glutámico, L-alanina, L-tirosina y L-lisina, parcialmente acetilados. No se conoce su mecanismo de acción, pero se ha sugerido que podría actuar como un péptido que mimetiza a la proteína base de la mielina, provocando un efecto inductor de los linfocitos T supresores, deficitarios en la EM, e inhibiendo el efecto de los antígenos anti-mielina del SNC, al inhibir el efecto de los linfocitos T autorreactivos. También se cree que actúa sobre las células dendríticas, que tienen una intensa capacidad presentadora de antígenos, orquestando las respuestas Th1 y Th2. Es capaz de reducir en un 30% el número de recaídas, y la discapacidad resultante, en los pacientes con EM de tipo remitente-recurrente. Sin embargo, no hay evidencia de que tenga efectos beneficiosos sobre la duración o la gravedad de la recaída, ni datos clínicos significativos en pacientes afectados con formas progresivas de la enfermedad.
Una opción en esas situaciones es natalizumab, un anticuerpo monoclonal que inhibe selectivamente las moléculas de adhesión, uniéndose a la subunidad α4 de las integrinas humanas, evitando así la penetración de los leucocitos al SNC inflamado y facilitando con ello la reducción de la inflamación y de las lesiones neurológicas asociadas a la EM. Natalizumab ha probado su capacidad de reducir la frecuencia de los ataques y frenar, en cierto grado, la progresión de la enfermedad a través de un mecanismo innovador (que le permite ser combinado con otras terapias). Tiene eficacia clínica contrastada (en estudios de 2 años de duración) en cuadros insatisfactoriamente tratados con IFN-β y una buena tolerabilidad general del tratamiento, con una incidencia global de eventos adversos solo levemente superior al placebo y sin los molestos síntomas de tipo gripal del IFN. Todo ello, añadido a una pauta de administración notablemente más aceptable, ha motivado que haya sido autorizado como tratamiento modificador de la enfermedad en monoterapia en la EM remitente-recurrente muy activa para pacientes con elevada actividad de la enfermedad a pesar del tratamiento con un IFN-β, o bien pacientes con EM remitente-recurrente grave de evolución rápida.
Más recientemente, la EMA autorizó el uso de cladribina (Mavenclad®) en adultos con EM recurrente muy activa. Este análogo de desoxiadenosina es más resistente que ésta a la degradación, lo cual aumenta su permanencia intracelular. Ejerce una acción selectiva sobre los linfocitos frente a otras células en la medula ósea; por tanto, tiene mayor efecto sobre las células del sistema inmunitario adaptativo que sobre el sistema innato: en los linfocitos, donde es eficazmente fosforilada a 2-clorodesoxiadenosina trifosfato, se acumula e induce la apoptosis al interferir la síntesis de ADN (Cuéllar, 2018).
Por otra parte, hasta hace poco el único abordaje eficaz de la EM progresiva primaria, que afecta aproximadamente al 15% de los pacientes, era la terapia inmunosupresora. Los fármacos inmunosupresores más utilizados son ciclofosfamida2, azatioprina y natalizumab. El fundamento de la aplicación de ciclofosfamida2 y azatioprina es la disminución de las células en rápida proliferación, entre ellas las linfoides, teóricamente responsables de la destrucción de la mielina en el SNC. Sin embargo, su uso en las formas progresivas más graves no ha mostrado un beneficio uniforme y tienen notables riesgos tóxicos. No obstante, azatioprina, administrada sola o junto con corticoides orales en dosis bajas, ha evidenciado una eficacia modesta en algunos aspectos clínicos como la rapidez de progresión o el número de recaídas, pero no en la incapacidad; este leve beneficio es el principal motivo que justifica que continúe siendo un fármaco usado en pacientes con múltiples brotes o en rápida progresión. Otros fármacos inmunosupresores ensayados en las formas progresivas primarias, con resultados más o menos decepcionantes (por su escasa eficacia o por su inaceptable toxicidad), han sido ciclosporina, clorambucilo y metotrexato.
Pero en 2019 se comercializó el primer fármaco con indicación específica en EM progresiva primaria, ocrelizumab, que también está aprobado para el tratamiento de pacientes adultos con formas recurrentes de EM y patología activa. Se trata de un anticuerpo monoclonal que actúa de forma selectiva contra linfocitos B que expresan CD20 (un antígeno de superficie celular que se encuentra en linfocitos pre-B, linfocitos B maduros y linfocitos B de memoria): su acción inmunomoludadora en EM parece deberse a la depleción de linfocitos B, limitando su efecto en la destrucción de las vainas de mielina neuronales (evento crucial en la patogénesis de EM). Reduce notablemente (47%) la tasa de recaídas en pacientes con EM recurrente, emergiendo como una nueva alternativa de segunda línea (como natalizumab, fingolimod o alemtuzumab) para aquellos pacientes que no respondan al menos a un FAME, y también disminuye un 24% el riesgo de progresión de la enfermedad en los pacientes con EMPP (Fernández-Moriano, 2019).
En España, las recomendaciones recogidas en el Documento del Grupo de Consenso de la Sociedad Española de Neurología (García-Merino et al., 2017) sobre el uso de medicamentos en EM orientan al uso de las siguientes opciones en el tratamiento inicial de la EM remitente-recurrente (la más común): INF-β, acetato de glatirámero, teriflunomida o dimetilfumarato. Natalizumab o fingolimod son considerados alternativas de tratamiento en aquellos casos de evolución rápida y agresiva. Para aquellos pacientes que no responden al tratamiento con inmunomoduladores, continúan presentando brotes y actividad lesional (evidenciada por técnicas de neuroimagen) o tienen formas muy graves de inicio, se valora la selección de natalizumab, fingolimod, ocrelizumab o de cladribina según factores dependientes del paciente (gravedad clínica, comorbilidades, etc.); alemtuzumab se suele reservar para pacientes no candidatos a natalizumab o fingolimod. Pero incluso a pesar del tratamiento con fármacos de 1ª línea, un número importante de pacientes continúa presentando brotes de la enfermedad y/o acumulando discapacidad. Teniendo en cuenta el amplio abanico de opciones terapéuticas disponibles, es indispensable conocer en profundidad el perfil de eficacia y seguridad de cada uno de ellos y ponerlos en contexto frente a las alternativas disponibles, con el fin de decidir cuál es la opción más adecuada en cada caso.
En relación al tratamiento de las formas de EM secundarias progresivas, solo se disponía hasta ahora de tres fármacos autorizados: el interferón-β1b, para los pacientes en recaída, y ocrelizumab o cladribina, en pacientes con enfermedad activa o muy activa. Por tanto, las opciones de tratamiento para los pacientes que entran en una fase de progresión secundaria son escasas y están restringidas a pacientes con actividad inflamatoria persistente (AEMPS, 2021).
Acción y mecanismo
Siponimod es un modulador del receptor de la esfingosina 1-fosfato (S1P)3, que se une selectivamente a dos receptores de la S1P –S1P1 y S1P5– (2 de los 5 receptores acoplados a proteínas G que se han descrito para esa molécula), y actúa como un antagonista funcional en los receptores S1P1 de los linfocitos, que se insensibilizan al efecto de la S1P. En base a ello, el medicamento ha sido autorizado para el tratamiento por vía oral (una dosis al día) de pacientes adultos con esclerosis múltiple secundaria progresiva (EMSP) con enfermedad activa definida por brotes o por características de imagen típicas de actividad inflamatoria.
Mediante la unión y antagonismo funcional de los receptores S1P1 en los linfocitos4 (se une a esos receptores con un patrón dosis-dependiente y promueve la internalización y degradación de los mismos), el fármaco previene la señal bioquímica mediada por S1P que induce la induce la salida de dichas células desde los órganos linfoides –ganglios linfáticos– y, en consecuencia, provoca una redistribución linfocitaria: se reduce la salida de los linfocitos de los órganos linfoides (quedan “secuestrados”) y, con ello, su infiltración patológica al sistema nervioso central. Así, reduce el riesgo de inflamación y lesiones en el tejido nervioso en los pacientes con esclerosis múltiple.
La justificación de su uso en el tratamiento de la EM se basa, pues, en los efectos farmacodinámicos ya conocidos para fingolimod, cabeza de serie del grupo farmacológico. Por tanto, como sugerían los datos experimentales para dicho fármaco, es posible que la acción moduladora de siponimod pueda no limitarse a los linfocitos, sino que afecte también a los receptores de S1P presentes en los astrocitos que rodean la vaina mielínica axónica, favoreciendo la aparición de efectos neuroprotectores y/o reparadores (Cuéllar, 2011).
Los estudios in vivo desarrollados durante su desarrollo pre-clínico y clínico han permitido probar que siponimod ejerce una reducción transitoria y reversible, dosis-dependiente, del recuento absoluto de linfocitos en sangre periférica incluso tras una sola dosis (en las 6 horas siguientes a administrar entre 0,3 y 10 mg en sujetos sanos), lo cual se correlacionaba con una reversión efectiva de los déficits neurológicos, con reducción de la inflamación (menor infiltración de macrófagos y activación de la microglía) y de la demielinización en modelos animales. Por su capacidad de atravesar la barrera hematoencefálica, los niveles de exposición al fármaco en cerebro eran unas 5 veces superior que la correspondiente en plasma (EMA, 2019). Se ha descrito que la administración diaria continuada de siponimod sigue reduciendo los niveles de linfocitos hasta un recuento mínimo (media de 0,56 células/nl) que corresponde al 20-30% del valor inicial en un paciente típico con EMSP, pudiendo mantenerse bajos mientras se mantiene la pauta. En la gran mayoría de pacientes con EMSP (90%), el recuento linfocitario periférico se recupera hasta valores normales en los 10 días siguientes a la interrupción del tratamiento, tras la cual pueden persistir ciertos efectos residuales de disminución hasta 3-4 semanas desde de la última dosis (AEMPS, 2020).
Aspecto moleculares
Siponimod existe en un complejo co-cristal con el ácido fumárico (2 moléculas de siponimod por cada una de ácido fumárico), siendo su nombre químico el de ácido (2E)-but-2-enedioico ácido 1-({4-[(1E)-N-{[4-ciclohexil-3-(trifluoro)fenil]metoxi}etanimidoil]-2-etilfenil}metil)azetidina-3-carboxílico, que se corresponde con la fórmula C62H74F6N4O10 y un peso molecular relativo de 1.149,29 g/mol. La sustancia activa se presenta en un polvo blanco o blanquecino, no higroscópico, que es insoluble en soluciones acuosas a un pH < 7 y muy ligeramente soluble a pH > 7,5 o en fluido intestinal simulado, no siendo tampoco muy soluble en solventes orgánicos. Es una molécula polimórfica, y también aquiral a pesar de contener un doble enlace con configuración E (el isómero termodinámicamente favorecido).
La estructura del cabeza de serie del grupo, fingolimod, guarda una marcada similitud con el de la esfingosina (tras el correspondiente proceso de fosforilización, el fosfato de fingolimod emula a la esfingosina-1-fosfato), lo cual permitió explicar su efecto sobre los receptores de S1P en la superficie de los linfocitos. En cambio, a pesar del paralelismo farmacológico entre ambos, siponimod difiere estructuralmente de fingolimod (Figura 2), pero fue descubierto a partir de la molécula de éste mediante el diseño y síntesis de una serie de derivados alquilamino tras el desarrollo de complejos estudios de relación estructura-actividad (Pan et al., 2013).
Eficacia y seguridad clínicas
La eficacia y seguridad clínicas de siponimod por vía oral han sido adecuadamente contrastadas en su indicación aprobada mediante un ensayo pivotal aleatorizado de fase 3 (A2304 o EXPAND), que fue un estudio determinado por el número de eventos y el tiempo de seguimiento, con un diseño doble ciego, multicéntrico y multinacional (292 centros en 31 países), de grupos paralelos y controlado por placebo en su parte principal (la fase de extensión fue abierta y no controlada).
Dicho estudio enroló a un total de 1.651 pacientes de 18 a 60 años que debían de tener historia previa de EM remitente-recurrente, haber entrado en una fase progresiva independiente de recaídas o brotes hasta 6 meses antes (evidencia de progresión en los 2 años previos, pero sin brotes o uso de corticoides en los 3 meses previos al reclutamiento), y tener una puntuación en la escala EDSS5 de entre 3,5 y 6,0. En cambio, se excluyeron pacientes con tratamiento inmunosupresor duradero, en tratamiento con inductores potentes CYP2C9, o que hubieran recibido fingolimod durante > 6 meses. Así, las características demográficas y clínicas basales de los pacientes estuvieron bien equilibradas entre los brazos del estudio, destacando las siguientes: mediana de edad de 49 años, 60% mujeres, 95% de raza blanca, mediana de tiempo desde el diagnóstico de EM de 11,7 años, mediana de tiempo de conversión a EMSP de 2,5 años, mediana de la EDSS de 6,0 puntos, el 64% de los pacientes no había tenido ningún brote en los 2 años previos al estudio, el 76% no presentaba lesiones realzadas con gadolinio en su resonancia magnética (RM) inicial y el 78% había recibido al menos un tratamiento previo para la EM, siendo los más frecuentes IFN-β1a (42%), IFN-β1b (27%) y glatirámero (27%).
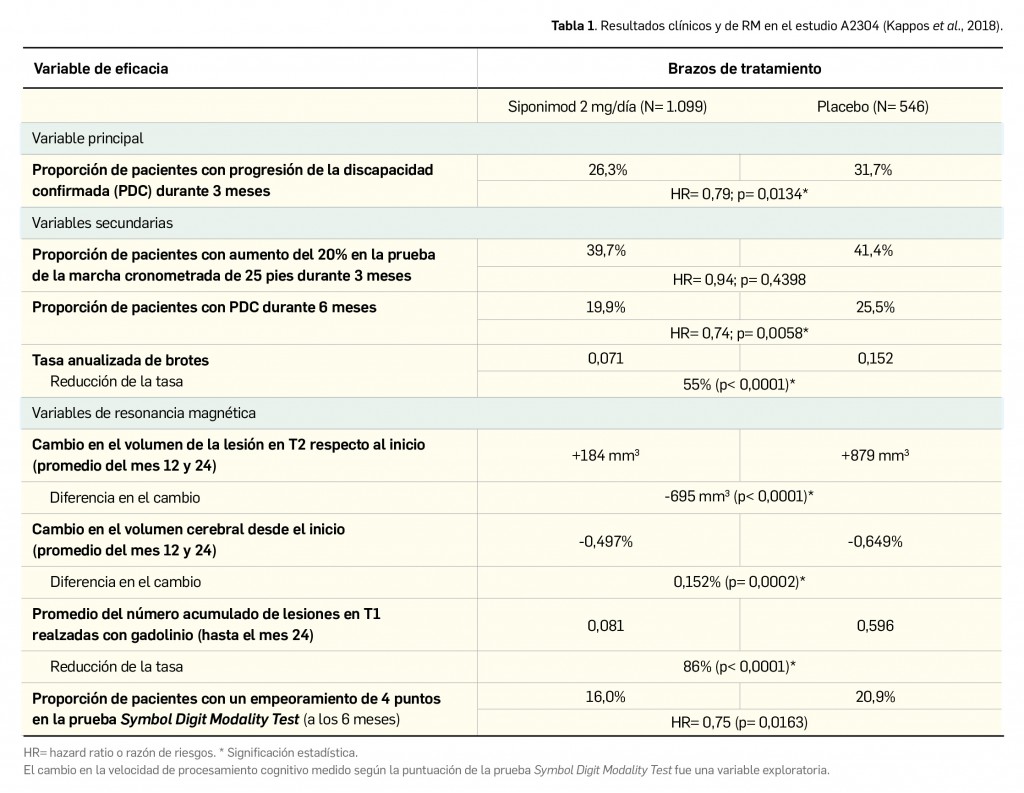
Los pacientes fueron asignados al azar (2:1) a recibir siponimod (N= 1.105) o placebo (N= 546) una vez al día, realizándose evaluaciones clínicas cada 3 meses y en el momento en que ocurría un brote, y de RM cada 12 meses. La duración del ensayo fue variable en función del paciente (media de 21 meses, rango: 1 día-37 meses); un 33% de los pacientes del grupo de siponimod y un 41% del grupo control discontinuó el tratamiento prematuramente durante la fase de doble ciego. Los investigadores consideraron como variable principal de eficacia el riesgo de progresión de la discapacidad confirmada (PDC)6 durante 3 meses y, entre las variables secundarias, destacan el riesgo de empeoramiento de ≥ 20% respecto al inicio –confirmado a los 3 meses– en la prueba de la marcha cronometrada de 25 pies y el cambio en el volumen de lesiones en T2.
Los resultados en la población general del estudio (Kappos et al., 2018) muestran que el fármaco retrasó de manera significativa la aparición de la PDC (Tabla 1). La mayoría de los pacientes no progresó durante el estudio, y la proporción de pacientes libres de PDC en el brazo de siponimod fue 82% al año (vs. 75% en el grupo placebo) y del 64% a los 3 años (vs. 56% con placebo). La reducción del riesgo de progresión fue variable pero consistente con siponimod en comparación con placebo en los subgrupos de pacientes evaluados, verificándose la eficacia con independencia del sexo, la edad, y diversos factores al inicio del estudio, como la frecuencia de recidivas, la duración de la EM y los grados de discapacidad. Sin embargo, los resultados reportados por los pacientes (por ejemplo, capacidad para caminar o impacto de la enfermedad en las actividades de la vida diaria o en la calidad de vida), incluidos como variables exploratorias, carecieron en su mayoría de significación estadística.
Cabe destacar que se llevó a cabo un análisis post-hoc en el subgrupo de pacientes con enfermedad activa (N= 778) –con brotes en los 2 años anteriores a la inclusión en el estudio y/o presencia de lesiones en T1 realzadas con gadolinio al inicio–, que representa a la población diana finalmente incluida en la indicación del medicamento. Estos pacientes presentaron características basales similares a las comentadas para la población global: mediana de edad de 47 años, puntuación de 6,0 en la escala EDSS y duración de la enfermedad de 15 años. En ellos, el tiempo hasta la aparición de PDC se retrasó significativamente con siponimod en comparación con placebo, tanto durante 3 meses (HR= 0,69; IC95% 0,53-0,91) como durante 6 meses (HR= 0,63; IC95% 0,47-0,86), y también se redujo en mayor medida la tasa de brotes confirmados (cociente de tasas= 0,54; IC95% 0,37-0,77). De igual modo, en la comparativa con placebo, las diferencias en el cambio en el volumen de la lesión en T2 (-1.163 mm3) y en el volumen cerebral (0,141%) fueron clínicamente relevantes a favor del fármaco. En el subgrupo de pacientes sin actividad al inicio (N= 827), sin embargo, sus efectos sobre la PDC durante 3 y 6 meses fueron inferiores respecto a la población general (reducciones del riesgo del 7% y del 13%), sin alcanzar significación estadística.
Por otra parte, la seguridad de siponimod a la dosis autorizada parece bien caracterizada en base a los datos derivados de 1.737 pacientes que han recibido al menos una dosis durante el desarrollo clínico, si bien solo un 7% fueron seguidos durante > 5 años, por lo que la seguridad a largo plazo no ha sido del todo esclarecida. La frecuencia global de eventos adversos es solo ligeramente superior a placebo (89,6% vs. 81,5%), siendo los más frecuentes las infecciones (49% en ambos grupos) y las alteraciones del sistema nervioso (39% vs. 32%); por su mayor incidencia con siponimod en comparación con placebo, destacan los siguientes: cefalea (15%), hipertensión (12%), mareo (7%), náuseas (7%), elevación de alanina aminotransferasa –ALT– (6%) y bradicardia (5%).
La mayoría de eventos adversos fueron leves-moderados en severidad, notificándose eventos adversos graves de grado ≥ 3 en un 17% de los pacientes tratados con siponimod (vs. 12% con placebo). Entre ellos, sobresale la mayor frecuencia de infecciones del tracto urinario, elevaciones de ALT y depresión. La tasa de discontinuación del tratamiento por problemas de seguridad fue del 8% con siponimod (vs. 5% con placebo), estando implicados fundamentalmente los siguientes eventos adversos: elevaciones de transaminasas, edema de mácula, bloqueo auriculo-ventricular, bradicardia y mareo. Ninguna de las muertes registradas en el ensayo pivotal se asoció al uso del fármaco.
Por su interés cualitativo, en base al mecanismo de acción y el perfil de seguridad de otros fármacos de la misma clase, se ha subrayado el riesgo de alteraciones cardiovasculares (sobre todo, bradicardia transitoria, retraso de la conducción auriculo-ventricular e hipertensión), que determinan su contraindicación/precaución de uso en ciertos pacientes cardiópatas y la necesidad de manejo del riesgo mediante un ajuste posológico inicial. También es importante destacar, también, que se debe evaluar el estado del genotipo de CYP2C9 antes de iniciar el tratamiento con siponimod, el cual está contraindicado en pacientes con genotipo CYP2C9*3*3 (actividad metabolizadora nula/lenta, en ≈0,4% de la población caucásica), debiéndose reducir la dosis también para heterocigotos con un solo alelo *3. Por último, tiene un importante potencial de involucrarse en interacciones farmacológicas, por lo que se recomienda consultar la Ficha Técnica del medicamento para una mayor información sobre las precauciones de su uso (AEMPS, 2020).
Aspectos innovadores
Siponimod es un modulador del receptor de la esfingosina 1-fosfato (S1P) que se une selectivamente a los receptores S1P1 y S1P5. Al actuar como un antagonista funcional en los receptores S1P1 de los linfocitos (induce de forma dosis-dependiente su internalización y degradación), que se insensibilizan al efecto de la S1P, el fármaco previene la salida de dichas células desde los órganos linfoides y, en consecuencia, provoca una redistribución linfocitaria: quedan “secuestrados” y se reduce su infiltración patológica al sistema nervioso central, limitando la inflamación y lesiones en el tejido nervioso. Además, reduce el recuento de linfocitos periféricos en un 20-30% respecto a niveles basales. En base a ello, el medicamento ha sido autorizado para el tratamiento por vía oral de pacientes adultos con esclerosis múltiple secundaria progresiva (EMSP) con enfermedad activa definida por brotes o por características de imagen típicas de actividad inflamatoria.
Los datos clínicos que han sustentado su autorización derivan de un único ensayo pivotal multicéntrico de fase 3 (EXPAND), con una fase principal doble ciego y controlada, en que el tratamiento diario con siponimod demostró superioridad clínica sobre placebo en pacientes con EMSP (N= 1.651). En esa comparativa, se observó un beneficio relevante con el uso del fármaco en la población global de pacientes, plasmado en el retraso de la progresión de la discapacidad, con una reducción relativa –estadísticamente significativa– del riesgo del 21% a los 3 meses y del 26% a los 6 meses, así como en una reducción del 55% en la tasa anualizada de brotes. La eficacia fue consistente en los subgrupos de pacientes analizados, con independencia de factores como sexo, edad o ciertas características clínicas basales, si bien el mayor beneficio clínico se constató en los pacientes con enfermedad activa al inicio (la población diana de la indicación aprobada). En ellos, siponimod redujo el riesgo de progresión de la discapacidad en un 31% a los 3 meses y en un 37% a los 6 meses en comparación con placebo, disminuyendo en un 46% la tasa de brotes confirmados de la enfermedad. En todo caso, el hecho de que los resultados reportados por los pacientes (por ejemplo, el impacto en la calidad de vida o en actividades de la vida diaria) carecieran de significación estadística a los 2 años plantea que la magnitud del beneficio clínico con el fármaco puede ser de limitada relevancia.
El conjunto de pacientes incluidos en el ensayo pivotal es representativo solo de un segmento de la población con ESMP, esto es, aquellos con enfermedad moderada (puntuación de EDSS basal de 6), edad intermedia (media de 49 años) y actividad inflamatoria moderada (≈50% de los pacientes tuvieron ≥ 1 recaída en los 2 años previos o lesiones captadoras de gadolinio al inicio), probablemente en fase precoz de la enfermedad. Esto no se alinea con las recomendaciones de la Guía Europea para la investigación clínica en EM, que recomienda incluir pacientes con EMSP sin recaídas o signos de inflamación activa a fin de probar convincentemente la eficacia en la prevención de la progresión de la discapacidad. La escasa proporción de pacientes que experimentó progresión de la discapacidad a los 6 meses tras 3 años de seguimiento en el estudio (30% con placebo y 25% con siponimod) contribuye a cuestionar la representatividad de la población incluida a pesar del amplio tamaño muestral (AEMPS, 2021).
Por otra parte, el perfil toxicológico de siponimod es similar al del otro miembro de su grupo farmacológico, fingolimod. Si bien la incidencia de eventos adversos es solo ligeramente superior a placebo, se trata de un perfil complejo, aún no bien caracterizado a largo plazo, en el que sobresalen los riesgos de seguridad referentes a la frecuencia de infecciones7 y a los eventos adversos cardiovasculares (bradicardia y bloqueo de la conducción cardiaca), lo que hace necesario un manejo específico en el seguimiento de los pacientes (monitorización hematológica, vacunación contra herpes zóster, ajuste de dosis, etc.). Además, por su frecuencia –superior a la notificada con placebo– destacan las alteraciones del sistema nervioso, cefalea e hipertensión, mientras que por su severidad se subraya el riesgo de infecciones del tracto urinario, elevaciones de ALT y depresión. En todo caso, la tasa de discontinuación durante el tratamiento no parece elevada (8% vs. 5% con placebo) y ninguna muerte se ha relacionado con el uso del fármaco.
Es preciso citar que en España se disponía hasta ahora de 3 opciones para el abordaje de pacientes con EMSP –interferón-β 1b, ocrelizumab y cladribina–, todos autorizados para el tratamiento de formas de EM recurrente, tanto EMRR como EMSP. A diferencia de ocrelizumab y cladribina, para siponimod sí se dispone de un estudio específico en pacientes con ESPM, de modo que la evidencia que avala su indicación, a pesar de las limitaciones comentadas, sería más robusta, como ocurre con interferón (que también demostró beneficio concluyente en pacientes con actividad inflamatoria). La ausencia de comparación directa con un tratamiento activo en el ensayo pivotal u otros estudios clínicos dificulta sustancialmente abordar el posicionamiento de siponimod en el arsenal terapéutico disponible. La comparación indirecta de los datos clínicos entre las cuatro opciones se ve también dificultada por la diferencia en los periodos de tratamientos, las variables evaluadas, etc.
En definitiva, se trata de un nuevo fármaco modificador de la enfermedad que aporta un beneficio clínico superior a placebo en pacientes con EMSP (sin que se pueda concluir de forma sólida sobre su beneficio en todo el espectro de pacientes), que media sus efectos inmunomoduladores mediante una redistribución linfocitaria: continúa la vía terapéutica inaugurada por fingolimod, y no aporta innovación a nivel mecanístico. El IPT de la AEMPS lo considera una alternativa a interferón-β 1b, ocrelizumab o cladribina en el tratamiento de pacientes con EMSP y actividad patológica, no siendo posible establecer la superioridad de uno sobre otro. Habida cuenta de la historia natural de la EM (las formas secundariamente progresivas se diagnostican tras el seguimiento de formas remitentes), los pacientes candidatos con EMSP habrán recibido uno o varios fármacos, según las guías de práctica clínica vigentes, pudiéndose cambiar de tratamiento cuando se confirme el predominio de la progresión: la elección deberá ser individualizada, valorando los tratamientos previos, las características del paciente y de la enfermedad, y el perfil de seguridad. Sin aportar ningún otro aspecto innovador reseñable respecto a las opciones disponibles (por ejemplo, vía de administración o mejor perfil toxicológico), la necesidad de realizar el genotipado del CYP2C9 antes de iniciar tratamiento puede condicionar el acceso a siponimod, pues se trata de una prueba que no se realiza de forma habitual en práctica clínica en todos los centros.
Valoración

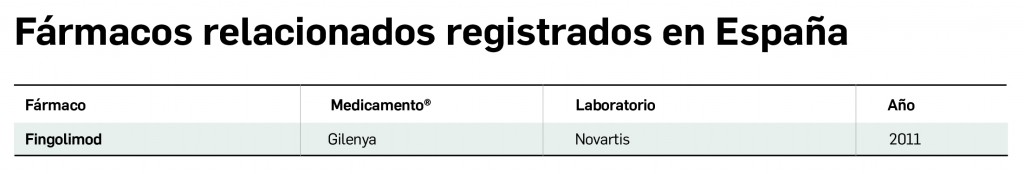
Fármacos relacionados registrados en España