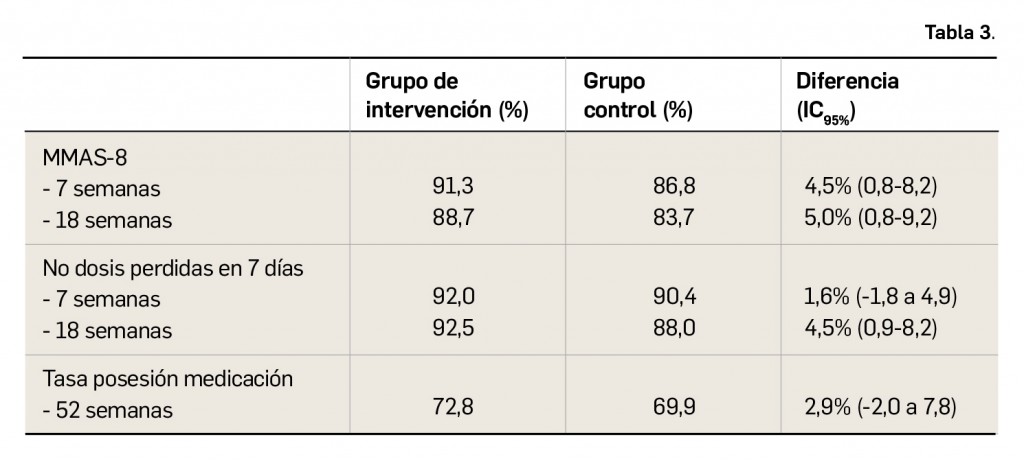
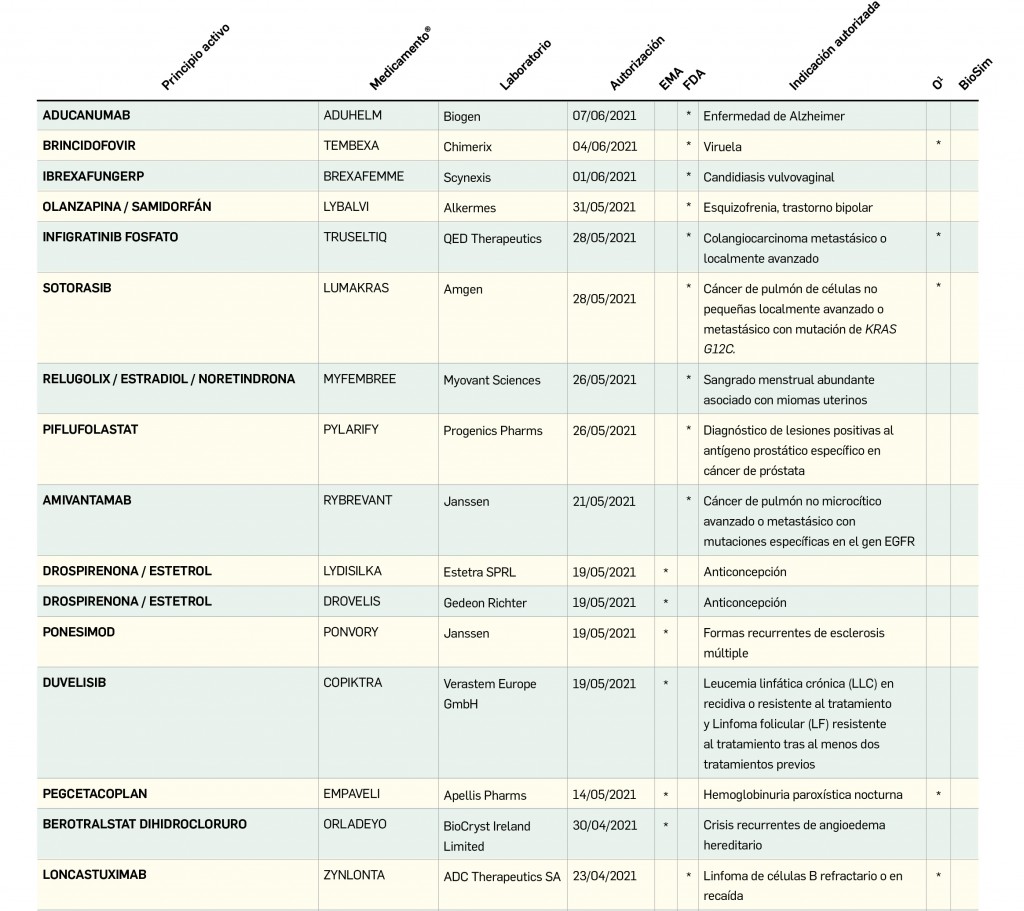
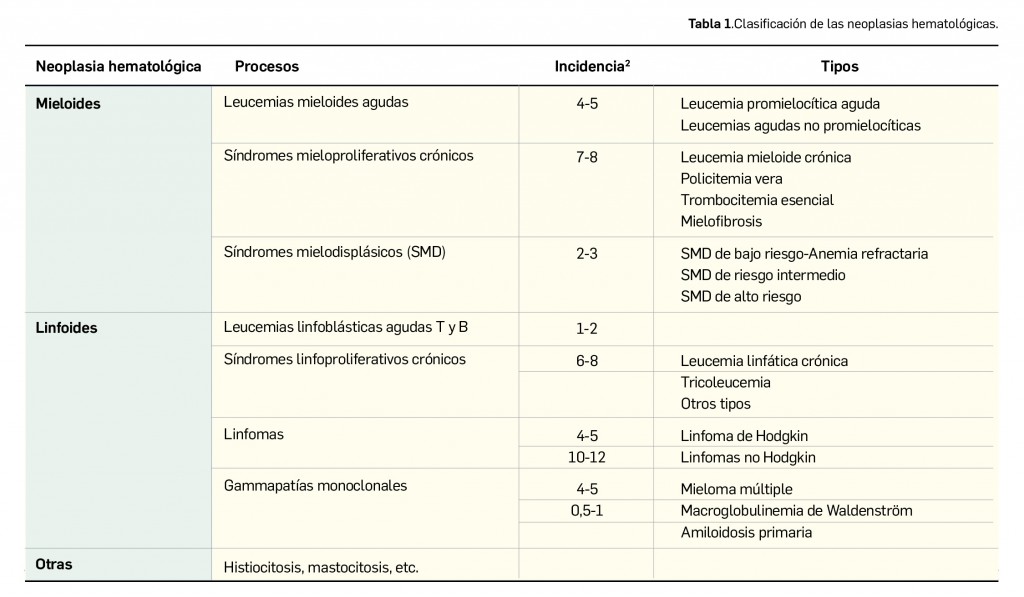
El subcomité para COVID-19 del Comité Asesor Mundial sobre Seguridad de las Vacunas (GACVS, por sus siglas en inglés) ha revisado los casos notificados de miocarditis asociados a las vacunas de ARNm, la de Pfizer (Comirnaty®) y la de Moderna (COVID-19 Vaccine Moderna®). Continúan las investigaciones globales con estudios en marcha para evaluar la asociación causal con estos efectos cardiacos.
El subcomité para COVID-19 del Comité Asesor Mundial sobre Seguridad de las Vacunas (GACVS, por sus siglas en inglés), publicó el 26 de mayo de 2021, el en el sitio web de la OMS información relacionada con los casos reportados de miocarditis tras la administración de vacunas frente a COVID-19 a base de ARNm, como las vacunas Comirnaty® (Pfizer-BioNTech) y la de Moderna (COVID-19 Vaccine Moderna®). A este respecto, indican que el 17 de mayo de 2021, el Grupo de Trabajo Técnico de Seguridad de las Vacunas frente COVID-19 del Comité Asesor sobre Prácticas de Inmunización (ACIP) de EE.UU. concluyó que son pocas las notificaciones de miocarditis hasta la fecha. Estos casos parecen ocurrir predominantemente en adolescentes y adultos jóvenes, más a menudo en hombres que en mujeres, y con mayor frecuencia dentro de los 4 días posteriores a la administración de la segunda dosis de la vacuna. La mayoría de los casos fueron leves y el seguimiento está en curso.
El subcomité del GACVS señaló que la mayor parte de la información recibida hasta ahora se basa en informes pasivos y espontáneos, y que se necesitan estudios más rigurosos que utilicen fuentes de datos alternativas y diseños de estudios más sólidos, incluida la comparación de poblaciones vacunadas y no vacunadas, para evaluar una posible asociación causal entre el evento y la vacuna. Algunos países, como Israel, Reino Unido y Estados Unidos, iniciarán este tipo de estudios.
El subcomité del GACVS subrayó la importancia de contar con una definición de caso armonizada, y que la Brighton Collaboration ha desarrollado recientemente un borrador de la definición de caso de miocarditis. En la UE, el PRAC también está revisando los casos notificados de miocarditis y pericarditis asociados a Comirnaty® y COVID-19 Vaccine Moderna®. Un caso de miocarditis ha ocurrido en España, asociado a Comirnaty®, y ya ha sido notificado y publicado. En Nueva Zelanda, se han notificado 2 casos de miocarditis y otros 2 casos de miopericarditis asociados a la inoculación de Comirnaty®, y su agencia reguladora de medicamentos (MedSafe) está evaluando todos los datos para confirmar su relación causal.
Recomendaciones
Si bien se reconocen los claros beneficios de las vacunas de ARNm en la reducción de muertes y hospitalizaciones debidas a la COVID-19, el subcomité GACVS alienta a todos los profesionales de la salud a informar sobre todos los eventos de miocarditis, pericarditis y endocarditis, así como otros eventos adversos, observados con estas y otras vacunas. El “Manual de vigilancia de la seguridad de la vacuna frente a COVID-19” de la OMS proporciona orientación a los países sobre el seguimiento de la seguridad y el intercambio de datos de eventos adversos para las nuevas vacunas frente a COVID-19.




 eciso subrayar que el componente CD3ζ es crítico para iniciar la activación de las células T y la actividad antitumoral (a través de un inmunorreceptor intracelular con un motivo de activación basado en tirosina), mientras que el dominio 4-1BB es responsable de aumentar la expansión y la persistencia de dichas células. Así, los receptores quiméricos CAR que portan los dominios de señalización CD3ζ son suficientes para desencadenar la activación y proliferación, pero no para impulsar la expansión in vivo y la persistencia de las células CAR-T. Por tanto, la adición del dominio de transducción intracelular 4-1BB (CD137) mejora su activación en comparación con los linfocitos que expresan receptores equivalentes que carecen del mismo: en modelos animales preclínicos, la inclusión del dominio de señalización 4-1BB incrementó significativamente la actividad antitumoral y la persistencia de los CAR en comparación con la inclusión del dominio de señalización CD3ζ solo.
eciso subrayar que el componente CD3ζ es crítico para iniciar la activación de las células T y la actividad antitumoral (a través de un inmunorreceptor intracelular con un motivo de activación basado en tirosina), mientras que el dominio 4-1BB es responsable de aumentar la expansión y la persistencia de dichas células. Así, los receptores quiméricos CAR que portan los dominios de señalización CD3ζ son suficientes para desencadenar la activación y proliferación, pero no para impulsar la expansión in vivo y la persistencia de las células CAR-T. Por tanto, la adición del dominio de transducción intracelular 4-1BB (CD137) mejora su activación en comparación con los linfocitos que expresan receptores equivalentes que carecen del mismo: en modelos animales preclínicos, la inclusión del dominio de señalización 4-1BB incrementó significativamente la actividad antitumoral y la persistencia de los CAR en comparación con la inclusión del dominio de señalización CD3ζ solo.


 Originaria de las zonas montañosas del suroeste de China e Himalaya, crece en Europa, Asia y Norteamérica, en zonas frías árticas y montañosas (Rusia, Escandinavia, etc.). En España se localiza en los Pirineos. Además, se cultiva en Europa y Norteamérica. Es una planta herbácea (Figura 1), perenne, que puede alcanzar hasta 30 o más cm de altura. Posee numerosos rizomas carnosos, densos, con escamas foliáceas, que se unen en su base a una raíz principal y desprenden cuando se cortan, un olor que recuerda al de la rosa. Dichos rizomas tienen la superficie de color pardo grisáceo, brillante y, una vez descortezados, se puede observar una capa color amarillo dorado. Los tallos, erectos, cilíndricos, no ramificados, llevan hojitas alternas, carnosas, cortamente pecioladas, glabras e irregularmente dentadas. Presenta inflorescencias cimosas terminales con flores amarillas (Figura 2); es una planta dioica, con flores masculinas y femeninas.
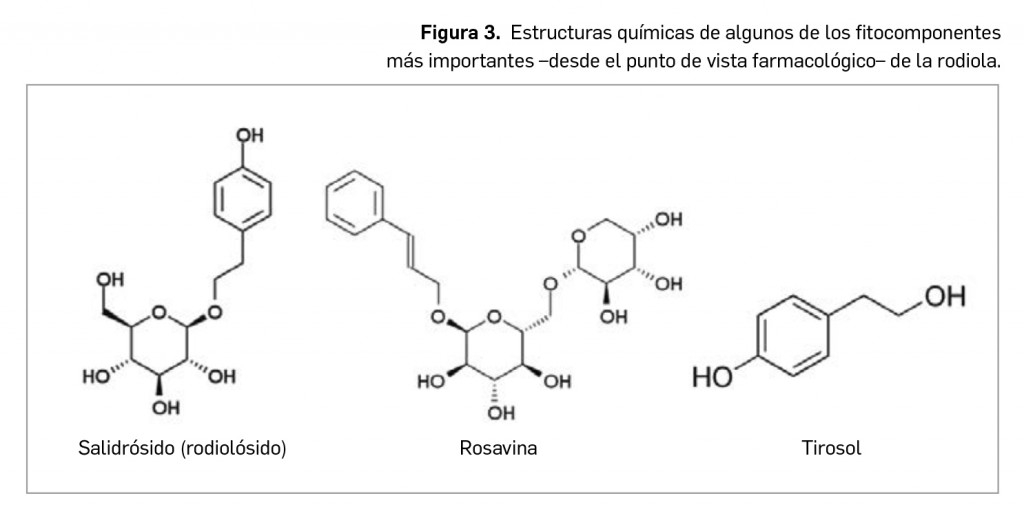
Originaria de las zonas montañosas del suroeste de China e Himalaya, crece en Europa, Asia y Norteamérica, en zonas frías árticas y montañosas (Rusia, Escandinavia, etc.). En España se localiza en los Pirineos. Además, se cultiva en Europa y Norteamérica. Es una planta herbácea (Figura 1), perenne, que puede alcanzar hasta 30 o más cm de altura. Posee numerosos rizomas carnosos, densos, con escamas foliáceas, que se unen en su base a una raíz principal y desprenden cuando se cortan, un olor que recuerda al de la rosa. Dichos rizomas tienen la superficie de color pardo grisáceo, brillante y, una vez descortezados, se puede observar una capa color amarillo dorado. Los tallos, erectos, cilíndricos, no ramificados, llevan hojitas alternas, carnosas, cortamente pecioladas, glabras e irregularmente dentadas. Presenta inflorescencias cimosas terminales con flores amarillas (Figura 2); es una planta dioica, con flores masculinas y femeninas. La parte empleada en terapéutica son las raíces y rizomas. La droga contiene un número elevado de principios activos (se han aislado más de cien), siendo mayoritarios los de tipo fenólico. Los principales (Figura 3) son: fenilpropanoides conocidos como rosavinas (rosavina, rosina, rosarina, etc.); derivados del feniletanol (salidrósido o rodiolósido, tirosol); monoterpenos (rosiridol, rosiridina); flavonoides (rodionina, rodiosina, rodiolina –este último es un flavonolignano–, etc.); ácidos fenólicos (ácido gálico y derivados, ácido clorogénico, hidroxicinámico, etc.); cumarinas; glucósidos cianógenos; y triterpenos (beta-sitosterol, daucosterol). Parece ser que el salidrósido se encuentra en todas las especies del género Rhodiola, mientras que las rosavinas son casi específicas de R. rosea. Los compuestos de mayor interés en lo que respecta a la actividad farmacológica de la droga son los fenilpropanoides y los derivados del feniletanol. Rosavinas y salidrósido se encuentran en una relación aproximada de 3:1.
La parte empleada en terapéutica son las raíces y rizomas. La droga contiene un número elevado de principios activos (se han aislado más de cien), siendo mayoritarios los de tipo fenólico. Los principales (Figura 3) son: fenilpropanoides conocidos como rosavinas (rosavina, rosina, rosarina, etc.); derivados del feniletanol (salidrósido o rodiolósido, tirosol); monoterpenos (rosiridol, rosiridina); flavonoides (rodionina, rodiosina, rodiolina –este último es un flavonolignano–, etc.); ácidos fenólicos (ácido gálico y derivados, ácido clorogénico, hidroxicinámico, etc.); cumarinas; glucósidos cianógenos; y triterpenos (beta-sitosterol, daucosterol). Parece ser que el salidrósido se encuentra en todas las especies del género Rhodiola, mientras que las rosavinas son casi específicas de R. rosea. Los compuestos de mayor interés en lo que respecta a la actividad farmacológica de la droga son los fenilpropanoides y los derivados del feniletanol. Rosavinas y salidrósido se encuentran en una relación aproximada de 3:1.