Queridos lectores,
Afrontamos el inicio del periodo estival recibiendo noticias esperanzadoras sobre la evolución de la vacunación en nuestro país, con cifras epidemiológicas paulatinamente decrecientes de la pandemia por COVID-19. En estas semanas, son diversos los temas relativos a dicho proceso vacunal los que copan la actualidad sanitaria. Desde la discutida suspensión temporal de las patentes de las vacunas hasta la influencia que, sobre la efectividad de los medicamentos autorizados, puede tener la expansión de distintas variantes virales –que, por cierto, han sido acertadamente rebautizadas por la OMS con letras del alfabeto griego, a fin de evitar la estigmatización de determinados territorios donde se identificaron por primera vez–. La adecuación de las vacunas a posibles variantes patógenas que puedan escapar a la inmunidad que aportan será otro reto científico apasionante, que tendrá que ser abordado en los próximos meses.
Abogamos desde estas líneas por el empujón final que nos acerque a ese deseado nivel (≥ 70%) de cobertura vacunal que permita iniciar la flexibilización de las medidas preventivas imperantes, para lo cual se hace necesario también asegurar la vacunación de los colectivos de profesionales sanitarios que pudieran permanecer sin inmunizar por completo, como ha sido el caso de algunos farmacéuticos en ciertas comunidades autónomas. Esta demanda se alinea con el dato arrojado por el último Eurobarómetro, que refleja que 7 de cada 10 españoles considera que la principal prioridad a nivel europeo debe ser la salud, para que todo lo demás pueda funcionar. Con tales objetivos, debemos, entre todos, combatir los posibles frenos a dicho proceso, tratando de evitar la adición de interrogantes desde organizaciones profesionales o autoridades sanitarias, como ha sido, por ejemplo, la recomendación de la pauta heteróloga de vacunación.
En otros campos de la ciencia, hemos asistido este mes a interesantes avances. La secuenciación por primera vez del genoma humano completo (con 3.055 millones de letras), que abre una nueva era en la genómica y en la investigación de posibles tratamientos para patologías como el cáncer, o la aprobación en EE.UU. de la primera terapia en dos décadas frente a la enfermedad de Alzheimer, que ha generado una amplia discusión en la comunidad científica, son algunas de las mejores muestras. No obstante, aunque haya mucha innovación en el campo de los medicamentos, si los pacientes no tienen acceso a ella en su ámbito asistencial, no se traducirá en un gran avance terapéutico. Esta es una cuestión a mejorar en nuestro sistema sanitario, toda vez que se constatan cifras como las divulgadas por la Federación Europea de Asociaciones de la Industria Farmacéutica (EFPIA), que indican que en España poco más de la mitad de los nuevos medicamentos aprobados en la UE en los últimos años son financiados y realmente accesibles para el paciente, por debajo de los principales países de nuestro entorno.
Con respecto a los contenidos de este nuevo número de PAM, destacaríamos una revisión monográfica inaugural sobre el cáncer renal, un tipo de tumor que representa el paradigma de patología oncológica en que el desarrollo de la investigación ha permitido un gran progreso de su terapéutica en los últimos años. También se incluye la evaluación de la innovación de tres medicamentos de reciente aprobación: una novedosa terapia CAR-T, que ha recibido una autorización de uso con cláusula de exención hospitalaria, y dos medicamentos con extractos alergénicos indicados en pacientes con alergia al veneno de himenópteros. Otros artículos en secciones como Vacunas, Actualidad Farmacoterapéutica o Farmacovigilancia complementan un abordaje integral de las novedades en el mundo del medicamento.
Deseamos que disfrutéis de su lectura y, dado el caso, también de las merecidas vacaciones de verano, sin olvidar, en estos días, la relevancia de la adecuada fotoprotección. Recibid un afectuoso saludo,
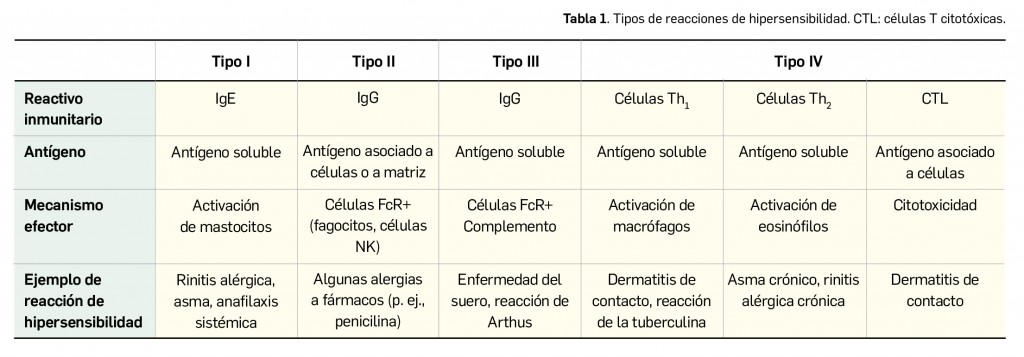
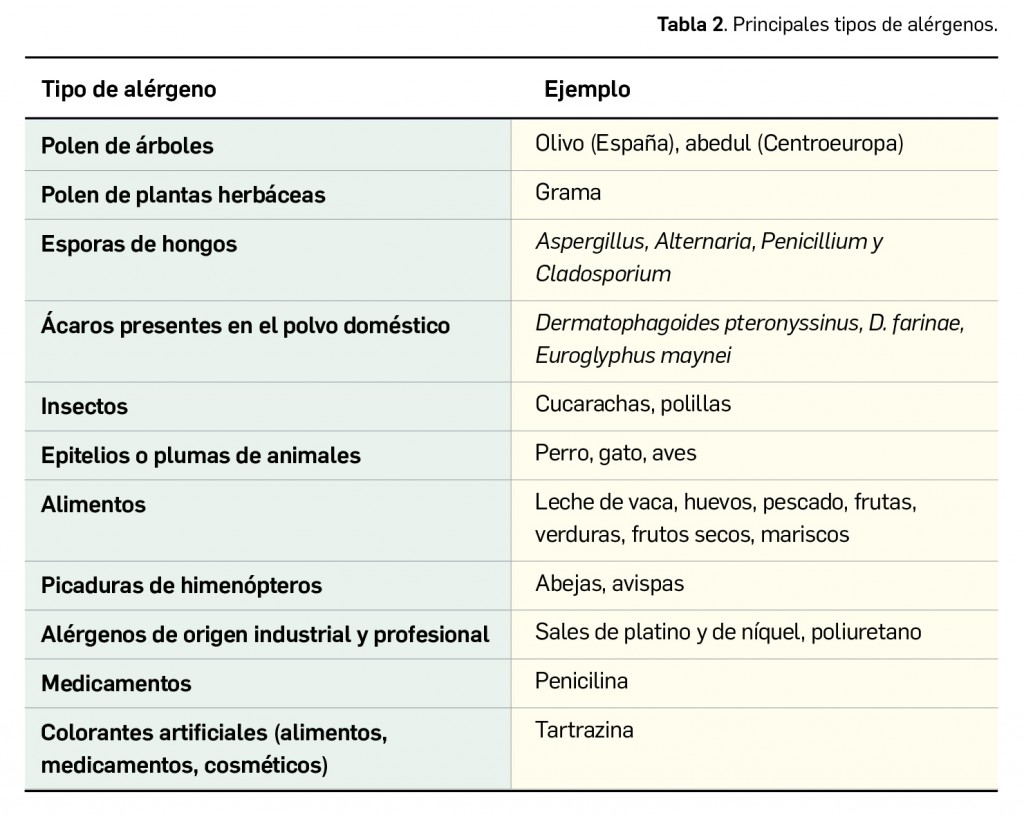
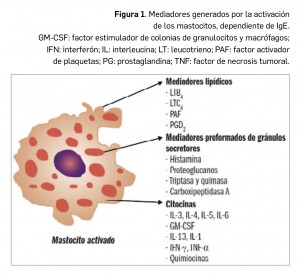 En definitiva, la alergia se considera a menudo como un concepto equivalente al de la hipersensibilidad de tipo I (reacciones de hipersensibilidad de tipo inmediato mediadas por IgE). La IgE es producida por las células plasmáticas localizadas en los ganglios linfáticos que drenan el sitio de entrada del antígeno o, localmente, en los sitios de las reacciones alérgicas por las células plasmáticas derivadas de los centros germinales en desarrollo en el tejido inflamado. La IgE difiere de otros isotipos de anticuerpo en que se localiza de forma predominante en los tejidos, donde se halla unida a los mastocitos por receptores de superficie de alta afinidad, denominados FcR1. La unión del antígeno a la IgE entrecruza estos receptores, lo que provoca la liberación de mediado-res químicos por parte de los mastocitos, que podrían conducir al desarrollo de una reacción de hipersensibilidad de tipo I.
En definitiva, la alergia se considera a menudo como un concepto equivalente al de la hipersensibilidad de tipo I (reacciones de hipersensibilidad de tipo inmediato mediadas por IgE). La IgE es producida por las células plasmáticas localizadas en los ganglios linfáticos que drenan el sitio de entrada del antígeno o, localmente, en los sitios de las reacciones alérgicas por las células plasmáticas derivadas de los centros germinales en desarrollo en el tejido inflamado. La IgE difiere de otros isotipos de anticuerpo en que se localiza de forma predominante en los tejidos, donde se halla unida a los mastocitos por receptores de superficie de alta afinidad, denominados FcR1. La unión del antígeno a la IgE entrecruza estos receptores, lo que provoca la liberación de mediado-res químicos por parte de los mastocitos, que podrían conducir al desarrollo de una reacción de hipersensibilidad de tipo I.