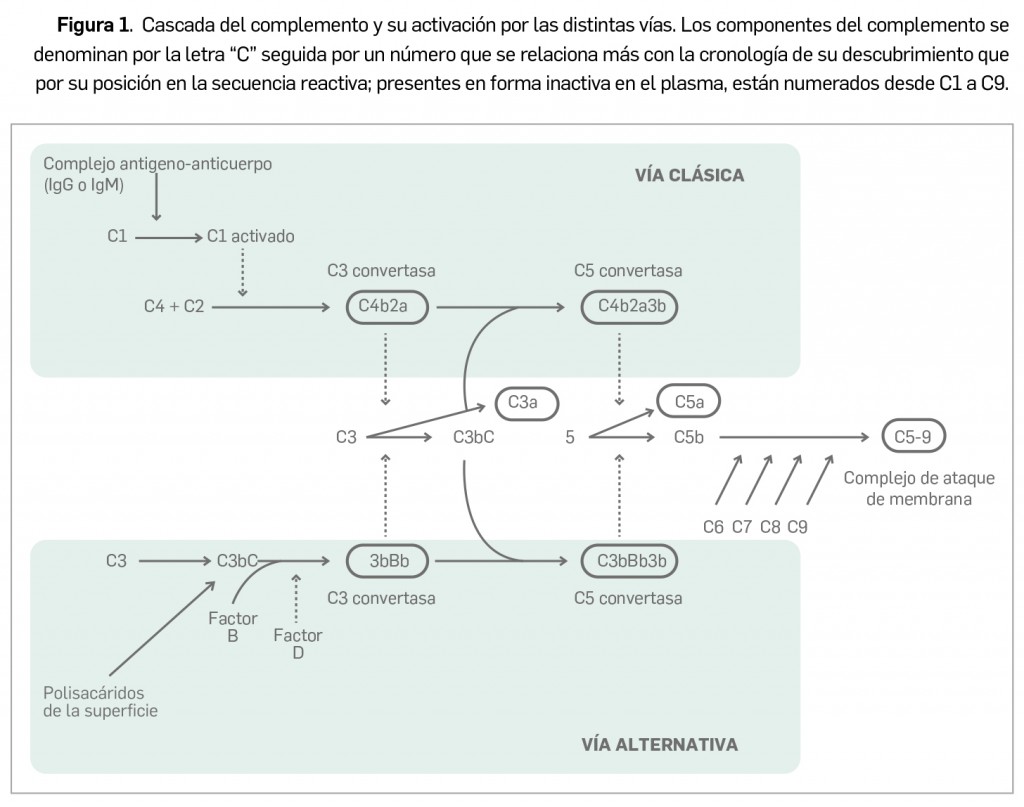
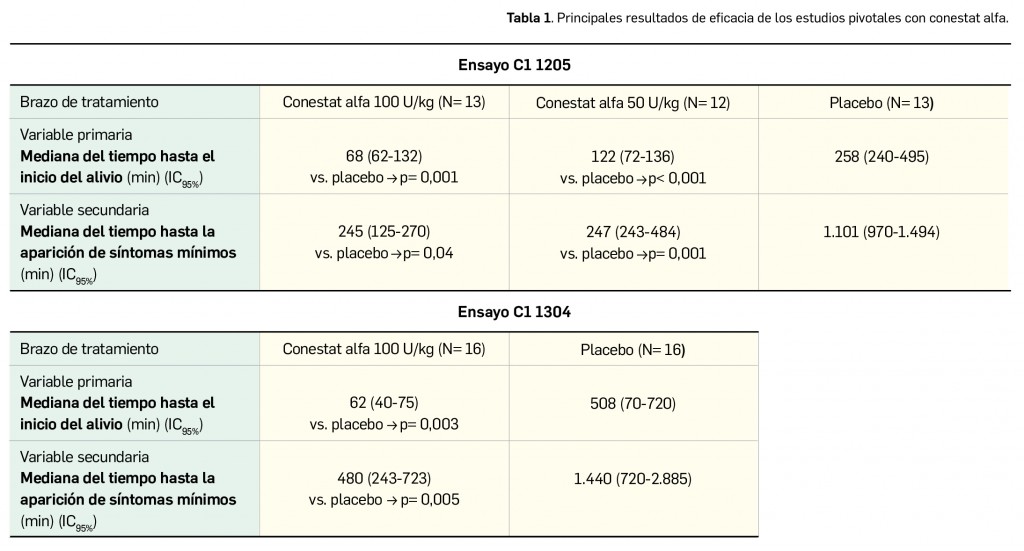
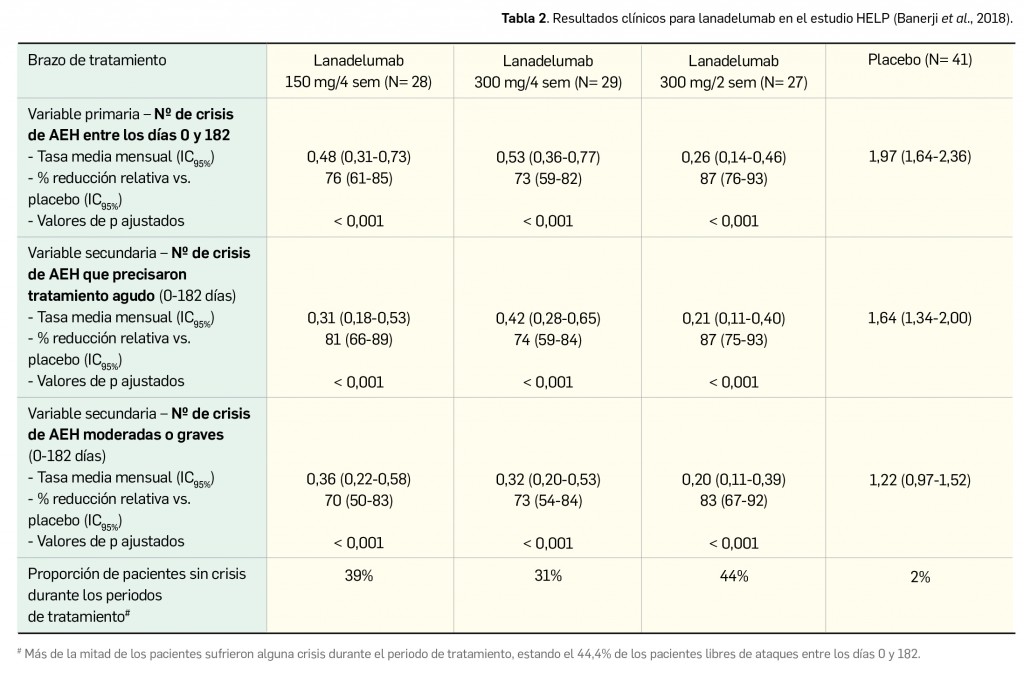
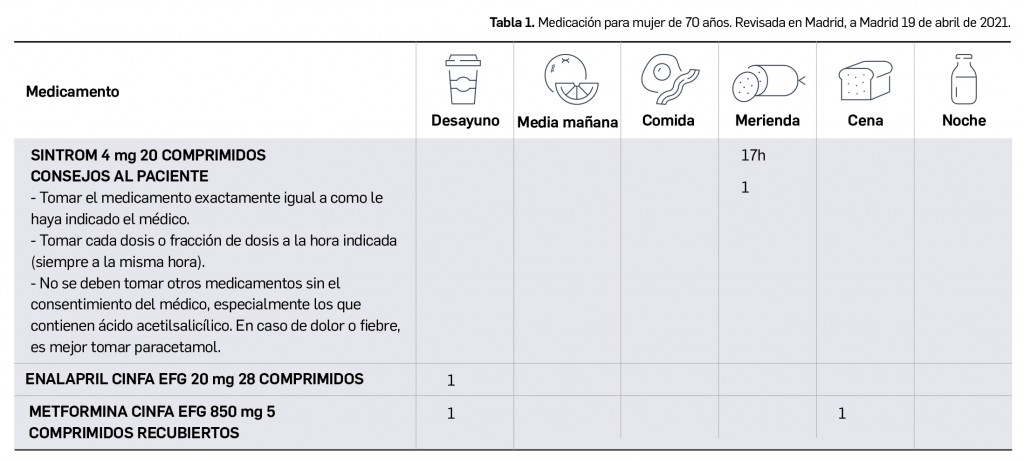
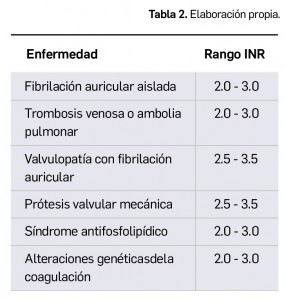
Según se ha comentado en números anteriores de Panorama Actual del Medicamento, han aparecido y siguen apareciendo –un proceso que previsiblemente continuará en un futuro próximo– distintas variantes virales del agente causal de la COVID-19 con mutaciones específicas en epítopos clave de la proteína S, aparecidas a partir del “virus original” aislado en los primeros casos de Wuhan que fue dominante a nivel global en los inicios de la pandemia. Una de las más importantes por su relevancia clínica es la variante B.1.1.7, popularmente conocida como variante británica, que se ha extendido ampliamente y es ya mayoritaria en numerosos países de Europa, incluido España.
Un reciente estudio de cohortes ha secuenciado y analizado las muestras positivas para SARS-CoV-2 (tras confirmación por PCR) recogidas de pacientes ingresados en dos hospitales de Londres desde el 9 de noviembre y hasta el 20 de diciembre de 2020. De los 496 pacientes que cumplieron los criterios de inclusión, solo se pudieron secuenciar adecuadamente muestras de 341. Los autores desarrollaron modelos de regresión de Poisson para determinar la relación entre la variante B.1.1.7 y la gravedad de la enfermedad, definida ésta como COVID-19 con una puntuación de ≥ 6 puntos en la escala de la OMS en los primeros 14 días de los síntomas o positividad. Realizaron, además, una serie de análisis genéticos suplementarios y de comparación de cargas virales en una cohorte de pacientes con infectividad persistente y otra de pacientes tratados con remdesivir.
Los resultados revelan que el 58% (198/341) de los pacientes tenían infección por la variante B.1.1.7, mientras que en el 42% (143/341) restante la infección se debía a otra variante viral. En la comparativa (B.1.1.7 vs. no-B.1.1.7), no se encontró evidencia de una asociación significativa entre la gravedad de la COVID-19 o la muerte por la enfermedad con la variante viral (ratio de prevalencia= 0,97; IC95% 0,72-1,31). Los análisis específicamente ajustados por hospital, sexo, edad, comorbilidades y etnia tampoco mostraron diferencias significativas (RP= 1,02; IC95% 0,76-1,38). En cambio, sí que se detectó una mayor carga viral en las muestras de pacientes infectados por la variante B.1.1.7 respecto a otras muestras con virus no-B.1.1.7, lo cual se dedujo a partir tanto del valor umbral de ciclo de PCR (p= 0,0085) como de la profundidad de lectura genómica (p= 0,0011). No se observaron mutaciones definitorias de la variante británica en 123 pacientes inmunodeprimidos con diseminación crónica ni en 32 pacientes tratados con remdesivir.
Así pues, se puede concluir que la evidencia emergente indica que la variante B.1.1.7 se asocia con una mayor transmisibilidad (por la mayor carga viral que implica), pero no parece asociarse con una mayor severidad de la COVID-19, al menos en los pacientes hospitalizados. Este tipo de trabajos contribuyen a un mayor conocimiento sobre las implicaciones clínicas de las nuevas variantes virales que van surgiendo, entre las que la variante india está tomando mayor protagonismo en las últimas semanas. La posibilidad de que determinadas mutaciones permitan que el virus escape de la inmunidad inducida por las vacunas es uno de los principales interrogantes de este fenómeno, y no ha sido aún sin esclarecido por completo.