Resumen
La vacuna monovalente AD26.COV2-S está compuesta por un vector de adenovirus humano tipo 26, recombinante y no replicativo, que contiene una secuencia de ADN codificante para la glicoproteína S del SARS-CoV-2 de longitud completa, en una conformación prefusión estabilizada mediante mutaciones específicas. La expresión transitoria de la proteína S por las células huésped en la zona de inyección permitirá que sea reconocida como antígeno extraño por las células del sistema inmunitario, desencadenando una respuesta adaptativa tanto de anticuerpos neutralizantes como de inmunidad celular por linfocitos T, que contribuirán a la protección frente a COVID-19 sintomática ante una futura exposición al SARS-CoV-2. En base a ello, el medicamento ha sido aprobado para la inmunización activa para prevenir la COVID-19 en personas de 18 años de edad y mayores en una pauta dosis única por vía intramuscular, debiéndose usar siempre siguiendo las recomendaciones oficiales.
Su autorización condicional se ha sustentado en los resultados del análisis de eficacia de un amplio estudio pivotal controlado de fase 3 (N > 44.000 adultos, casi 21.900 recibieron la vacuna) realizado en EE.UU., Latinoamérica y Sudáfrica. Con un seguimiento de unos dos meses, tras la confirmación de 116 casos de COVID-19 sintomática en el grupo de la vacuna y 348 casos en el grupo placebo, se determinó una eficacia protectora global en sujetos seronegativos para el SARS-CoV-2 del 66,9% tras 14 días desde la vacunación. Esa protección se mantuvo en el 66,1% en los casos de inicio después de ≥ 28 días. La eficacia se mostró independiente de factores como el sexo, la presencia de comorbilidades, e incluso de la edad, aportando una inmunoprotección superior en sujetos de mayor edad: tras ≥ 28 días, la eficacia vacunal fue del 65% entre 18 y 64 años y de hasta el 74% en participantes de ≥ 65 años. Además, un análisis exploratorio por regiones geográficas sugirió que su eficacia, aunque ligeramente menor, se mantiene en niveles elevados frente a las variantes virales B.1.351 y P.2, con una protección del 64% en Sudáfrica, el 68% en Brasil y el 72% en EE.UU. De modo interesante, la eficacia frente a la enfermedad grave fue de > 85% tras 28 días desde la vacunación, no registrándose ninguna muerte por COVID-19 entre las personas vacunadas; ese efecto fue consistente entre regiones geográficas. Persisten incertidumbres en torno a la eficacia de la vacuna en mayores de 75 años, la prevención de ingreso en UCI o mortalidad, frente a la infección asintomática y la capacidad de transmitir la enfermedad o sobre el tiempo de inmunidad conferida. Con respecto a la seguridad, se trata de una vacuna bien tolerada a corto plazo: el perfil toxicológico se define por reacciones adversas de gravedad leve-moderada y transitorias, destacando el dolor en el punto de inyección, la cefalea, la fatiga, mialgias y náuseas. Habrá que esperar a los datos tras el seguimiento de 2 años previsto en el ensayo pivotal para caracterizar su seguridad a medio-largo plazo, no pudiéndose descartar aún la posibilidad de anafilaxia ni de aparición de eventos trombóticos raros.
En definitiva, en el actual contexto de pandemia, con la aparición de nuevas variantes virales y la necesidad de disponer de suficientes medicamentos profilácticos autorizados, esta vacuna es la primera que se autoriza en pauta de 1 dosis, representando un cierto grado de avance terapéutico en su indicación, si bien no supone una innovación farmacológica disruptiva (el uso de vectores adenovirales no es una estrategia tan novedosa como lo fue el ARNm). Con un perfil de seguridad similar al resto de vacunas aprobadas en la UE, parece que aporta una inmunoprotección inferior a la de las vacunas de ARNm, que se mueven en tasas de eficacia del 94-95%, pero similar o superior a ChAdOx1-S (Vaxzevria®), cercana al 60%. Además, el grado de prevención de COVID-19 grave (> 85%) es muy relevante clínicamente. Contribuirá a ampliar el arsenal terapéutico para elevar las coberturas vacunales, aportando como ventaja –respecto a las de ARNm– su mayor estabilidad: no requiere congelación y se puede conservar 3 meses en nevera (2-8ºC) e incluso 12 horas a temperatura ambiente.
Aspectos fisiopatológicos
La enfermedad causada por coronavirus o COVID-19 –acrónimo derivado del inglés coronavirus disease 2019 (así designada porque inicialmente el virus se denominó “2019 novel coronavirus” o “2019-nCoV”)– es una patología infecciosa causada por el virus zoonótico denominado SARS-CoV-2. Este virus emergió por primera vez como un patógeno humano en China a finales de 2019, cuando en la ciudad de Wuhan (provincia de Hubei) las autoridades sanitarias locales informaron de un grupo de 27 casos de neumonía de etiología desconocida (incluyendo 7 casos graves), con una exposición común a un mercado mayorista de marisco, pescado y animales vivos en esa ciudad. El inicio de los síntomas del primer caso oficialmente reconocido fue el 8 de diciembre de 2019, y el 7 de enero de 2020 las autoridades chinas identificaron como agente causante del brote al nuevo tipo de virus de la familia Coronaviridae, cuya secuencia genética se hizo pública rápidamente, el 12 de enero. Los análisis filogenéticos revelaron posteriormente una relación más estrecha del citado virus con el coronavirus del síndrome respiratorio agudo severo (SARS-CoV; homología en torno al 79%) que con otros coronavirus que infectan a los humanos, incluido el coronavirus del síndrome respiratorio del Medio Oriente (MERS-CoV; en torno al 50%).
Desde entonces, hemos asistido a su rápida expansión –en mayor o menor grado– por la práctica totalidad de países del mundo, debido a su relativamente fácil transmisión persona-persona por vía aérea a través de secreciones respiratorias (gotículas emitidas al toser o estornudar) y aerosoles (generados incluso al hablar). El 11 de marzo de 2020 el brote llegó a ser calificado como pandemia por la Organización Mundial de la Salud.
Si bien son cifras constantemente cambiantes, según datos del Ministerio de Sanidad, a día 30 de abril de 2021 se ha registrado de forma oficial el diagnóstico confirmado de más de 148,3 millones de casos de COVID-19 a nivel mundial, de los cuales más de 49,9 millones se han notificado en Europa y unos 3,5 millones de casos en España. En términos de mortalidad, se ha estimado una tasa de letalidad variable entre países, que, por ejemplo, en países europeos oscila entre el 0,6% y el 3,0% (en España se ha estimado en el 2,2%). Hasta la fecha citada, se han reconocido más de 3,1 millones de muertes en todo el mundo motivadas por COVID-19. En el momento de redacción de este artículo, y a pesar de los intentos sin precedentes de control por los diferentes países, los casos confirmados de infección y fallecimiento por la enfermedad continúan aumentando a nivel mundial, y también en España. Se trata de una pandemia global que se ha convertido en el principal desafío para la salud pública y la estabilidad socioeconómica en muchas décadas.
El SARS-CoV-2 es un patógeno nuevo frente al cual no existe inmunidad preexistente y todas las personas están en riesgo de infección, en mayor medida aquellas con mayor riesgo de exposición al virus, como pueden ser los profesionales sanitarios u otros profesionales “de primera línea” en contacto con muchas personas. Se ha demostrado que, después de la infección, una proporción amplia de individuos –pero no la totalidad– desarrollan inmunidad protectora en términos de respuestas de anticuerpos neutralizantes e inmunidad mediada por células T. No obstante, actualmente no se conoce con seguridad en qué medida y durante cuánto tiempo se mantiene esta protección (se cree que puede perdurar más allá de 6 meses). Según la OMS, el 80% de las personas infectadas se recuperan sin necesidad de atención hospitalaria, mientras que el 15% desarrolla una enfermedad moderada-grave que sí requiere esa asistencia especializada, y el 5% necesita cuidados intensivos.
Hay consenso en la comunidad científica en torno al hecho de que la gran mayoría de infecciones cursan como casos asintomáticos (aspecto fundamental a la hora de tratar de contener la transmisión del virus) o enfermedad levemente sintomática por infección del tracto respiratorio superior, con manifestaciones que pueden asemejarse a las del catarro común, resultando en una completa recuperación clínica de los pacientes en menos de 2 semanas. Sin embargo, la presencia de otras patologías subyacentes tales como hipertensión, diabetes, enfermedad cardiovascular, patologías crónicas respiratorias o renales, obesidad, cáncer y otros estados de inmunodepresión (por ejemplo, pacientes trasplantados) se consideran, junto a una edad avanzada1, factores de riesgo para el desarrollo de una enfermedad grave y de mortalidad por COVID-19.
Aunque se ha descrito una amplia variabilidad de presentaciones clínicas de la COVID-19, se acepta que, cuando ésta es sintomática, la forma más común es aquella que se manifiesta con tos y fiebre, y la radiografía de tórax muestra opacidades “en vidrio esmerilado” o sombras irregulares, posiblemente de forma bilateral. Los síntomas más comunes en pacientes hospitalizados (en orden de mayor a menor frecuencia), posiblemente variables con el tiempo, incluyen: fiebre, tos seca, dificultad para respirar, fatiga, mialgias, náuseas/vómitos o diarrea, dolor de cabeza, debilidad y rinorrea. La anosmia (pérdida del olfato) o la ageusia (pérdida del gusto) pueden ser la única manifestación en aproximadamente el 3% de las personas que padecen COVID-19. En este sentido, los Centros para el Control y la Prevención de Enfermedades de EE.UU. han definido la enfermedad como la presencia de uno o más de los siguientes síntomas: fiebre, tos de nueva aparición o aumentada, falta de aire nueva o aumentada, escalofríos, dolor muscular nuevo o aumentado, pérdida del gusto u olfato, dolor de garganta, diarrea, vómitos, fatiga, cefalea, congestión o secreción nasal y/o náuseas.
La progresión de la enfermedad puede conducir a una de las mayores complicaciones, el síndrome de distrés respiratorio agudo (SDRA), que se manifiesta con disnea y fallo respiratorio agudo –suele requerir ventilación mecánica– y es susceptible de conducir hasta la insuficiencia multiorgánica y la muerte. Por otro lado, además de las secuelas respiratorias, la COVID-19 se ha relacionado con secuelas cardiovasculares (entre otras, daño miocárdico, arritmias, insuficiencia cardiaca y eventos tromboembólicos, incluyendo ictus isquémico), daño renal agudo (que puede hacer necesario un trasplante o diálisis) o complicaciones neurológicas (por ejemplo, encefalopatía).
Se ha estimado que la mediana del periodo de incubación tras la infección inicial es de 4 o 5 días hasta el desarrollo de los síntomas, si bien la mayoría de los pacientes sintomáticos experimentan síntomas entre los 2 y los 7 días posteriores a la infección, y prácticamente todos antes del día 12. Los pacientes con enfermedad grave o crítica pueden tardar una media de 3 a 6 semanas en recuperarse, habiéndose descrito, en los casos de fallecidos, un tiempo desde el inicio de los síntomas hasta la muerte variable entre 2 y 8 semanas. Un tiempo elevado de protombina o niveles anormalmente altos de proteína C reactiva son algunos de los biomarcadores que se han relacionado con la mayor gravedad de la enfermedad al ingreso hospitalario y con la necesidad de ingreso en UCI.
En una amplia proporción de las situaciones clínicas, y preferiblemente a los pocos días de la exposición (cuando debutan los síntomas), para el diagnóstico definitivo de infección activa por SARS-CoV-2 se emplea como método de referencia o gold standard la técnica molecular denominada reacción en cadena de la polimerasa con transcripción reversa (RT-PCR), basada en la detección del ARN viral; a día de hoy, la práctica general implica la toma de muestras mediante un hisopo o torunda en las fosas nasales, aunque también puede hacerse en la mucosa bucofaríngea (y se están empezando utilizar muestras de saliva). Sin embargo, la capacidad de esta técnica para determinar la duración de la infectividad de los pacientes es muy limitada.
Desde el punto de vista de la terapéutica, el manejo de la COVID-19 se ha desarrollado por completo durante el año 2020 y principios del 2021, a medida que ha ido avanzando el conocimiento biomédico sobre el virus y la enfermedad, tal y como se ha venido comentando en detalle en números previos de Panorama Actual del Medicamento.
Han sido numerosos los fármacos potencialmente candidatos que se han investigado en ensayos clínicos y de los cuales se ha descartado la eficacia terapéutica (por ejemplo, hidroxicloroquina, azitromicina o lopinavir/ritonavir). Actualmente, los dos únicos fármacos autorizados en la Unión Europea para el tratamiento de los casos de COVID-19 grave –que al menos requieren terapia de oxígeno suplementaria– son el antiviral remdesivir y el corticosteroide dexametasona, que han demostrado la capacidad de reducir significativamente el tiempo hasta la recuperación clínica y la mortalidad, respectivamente, en determinados subgrupos de pacientes. También se ha probado recientemente el beneficio sobre la mortalidad de tocilizumab en pacientes con enfermedad grave, fundamentado –como en el caso de la dexametasona– en sus propiedades antiinflamatorias, que atenuarían el proceso hiperinflamatorio a nivel pulmonar.
En todo caso, el tratamiento de los casos graves de COVID-19 a nivel hospitalario ha recurrido frecuentemente, además de al tratamiento sintomático de soporte (fluidoterapia, oxígeno, antitérmicos, antibióticos, etc.), al uso off label y, en muchos casos, al uso compasivo de otras opciones terapéuticas, entre las que sobresalen: plasma hiperinmune (con anticuerpos de pacientes que han superado la enfermedad), de inmunoglobulinas específicas (por ejemplo, banlanivimab, casirivimab, imdevimab2), estatinas, otros corticoides antiinflamatorios o agentes inmunomoduladores dirigidos (como el inhibidor de enzimas JAK baricitinib) y anticoagulantes. Estas terapias, algunas de las cuales han sido autorizadas en otros países y otras continúan en investigación, han mostrado un impacto variable y limitado sobre la gravedad y duración de la COVID-19, con diferentes tasas de eficacia según el estadio de la infección y las manifestaciones de la enfermedad. En la UE, no se dispone aún de tratamientos específicos autorizados para casos leves o moderados en pacientes no hospitalizados.
En resumen, aunque el tratamiento de la COVID-19 ha mejorado con la experiencia clínica, y teniendo en cuenta que aún hay cantidad de ensayos en marcha con diversos fármacos, algunos de los cuales están revelando resultados preliminares prometedores (plitidepsina, colchicina, molnupiravir, sarilumab, etc.), durante la pandemia en curso sigue existiendo una necesidad médica urgente y no cubierta de una prevención farmacológica eficaz, tanto para la protección de grupos particularmente vulnerables como para mitigar en un futuro las consecuencias sociales, sanitarias y económicas derivadas de las estrictas medidas necesarias para reducir la propagación del virus. Con la aprobación y uso de las vacunas, entre ellas la que aquí se comentará, se abre una puerta a la esperanza (EMA, 2021). Conviene subrayar que previamente se han autorizado –y comenzado a distribuir– otras tres vacunas frente a la COVID-19, entre ellas dos que fueron pioneras en su campo e incorporaron una innovación farmacológica disruptiva por tratarse de las primeras vacunas de uso humano basadas en ARNm (Fernández-Moriano, 2021a).
Finalmente, conviene hacer referencia a algunas de las principales características del agente causal de la COVID-19. En conjunto, los coronavirus son un grupo diverso de virus de ARN monocatenario y de sentido positivo, envueltos, que pertenecen a dos subfamilias –Coronavirinae y Torovirinae– de la familia Coronaviridae. Se descubrieron por primera vez en la década de 1960 y se clasifican, en función de sus relaciones filogenéticas y estructura genómica, en 4 géneros principales: Alphacoronavirus, Betacoronavirus, Gammacoronavirus y Deltacoronavirus. Actualmente, se conocen 6 coronavirus que infectan a humanos, además del SARS-CoV-2 recientemente identificado, a saber: HCoV-229E, HCoV-OC43, HCoV-NL63, HCoV-HKU1, SARS-CoV y MERS-CoV.
Encuadrado taxonómicamente en la subfamilia Betacoronavirus, el SARS-CoV-2 es, pues, un virus de ARN monocatenario (+ssRNA, por sus siglas en inglés) y envuelto, de unos 50-200 nm de diámetro cada virión, cuya secuencia genómica de referencia –de las primeras muestras secuenciadas3– está compuesta por 29.903 nucléotidos y presenta una estructura y orden de los genes similar a otros coronavirus. En dicho genoma, el gen ORF1ab codifica una poliproteína que se divide en proteínas no estructurales; tras él, se encuentran una serie de genes que codifican para las 4 proteínas estructurales del virus: S (espícula), E (envoltura), M (membrana) y N (nucleocápside); de todos ellos, el gen codificante para la proteína S es el más largo, con 3.822 nucleótidos. Aunque aún no está del todo claro su origen, los estudios filogenéticos realizados hasta la fecha sugieren que muy probablemente el SARS-CoV-2 provenga de murciélagos, y que de allí haya pasado al ser humano a través de mutaciones o recombinaciones sufridas en un hospedador intermediario, probablemente algún animal vivo del mercado de Wuhan (Cyranoski, 2020); se planteó que ese animal pudiera ser el pangolín, bien de forma directa o indirecta (a través de otra especie), sin que se haya llegado todavía a una conclusión definitiva.
De entre sus componentes, la proteína S o de la espícula (spike) del SARS-CoV-2 es la más relevante en la fisiopatología de la enfermedad. A través de ella –concretamente, de la subunidad S1– los viriones se unen a la enzima convertidora de angiotensina 2 (ECA-2) de las células hospedadoras humanas, que actúa como receptor principalmente en el tracto respiratorio, permitiendo –mediante la subunidad S2– la posterior fusión con la membrana celular (en un proceso en que también participa la proteasa celular TMPRSS2). La proteína S4 es una proteína trimérica de fusión (clase I) que existe en una conformación metaestable de prefusión antes de unirse a la célula diana, y contiene también un sitio de escisión polibásico, característica típicamente relacionada con un aumento de la patogenicidad y la transmisibilidad en otros virus. Por todo ello, se considera el antígeno más relevante en el desarrollo de vacunas, habiéndose probado que los anticuerpos dirigidos contra ella neutralizan el virus y provocan una respuesta inmunitaria que previene la infección en animales (Hoffman et al., 2020; Yan et al., 2020).
Una información mucho más extensa y detallada sobre la COVID-19 y el patógeno causal puede encontrarse en la Información Científica-Técnica5 publicada y periódicamente actualizada por el Ministerio de Sanidad, en base al nuevo conocimiento que va estando disponible. Además, se recomienda consultar el artículo monográfico sobre la COVID-19 publicado en la sección “Revisión” del número 442 de Panorama Actual del Medicamento, donde se hace referencia a la información microbiológica, los datos epidemiológicos actualizados y los aspectos clínicos, profundizando en los avances farmacoterapéuticos.
Acción y mecanismo
El nuevo fármaco es una vacuna monovalente compuesta por un vector viral único de adenovirus tipo 26 humano (Ad26) no replicativo, con un ADN que codifica para la glicoproteína S del SARS CoV 2, de longitud completa y en una conformación estabilizada. En base a ello, el medicamento ha sido autorizado para la inmunización activa para prevenir la COVID-19 causada por SARS-CoV-2, en personas de 18 años de edad y mayores. Se administra en una pauta de dosis única (de al menos 5×1010 partículas virales, que se corresponden con 8,92 log10 unidades infecciosas en 0,5 ml) por vía intramuscular, preferiblemente en el músculo deltoides en la parte superior del brazo, debiéndose usar siempre siguiendo las recomendaciones oficiales.
Su mecanismo de acción es similar al descrito para Vaxzevria®, la vacuna de AstraZeneca, y podría resumirse según lo descrito a continuación. Tras su administración por vía intramuscular, el vector adenoviral “infecta” (transfecta) las células huésped de la zona de la inyección, donde libera el ADN que conducirá a la expresión transitoria de la glicoproteína S del SARS CoV 2: el ADN será transcrito a ARNm en el núcleo celular y, posteriormente, traducido a la proteína en el ribosoma citoplasmático. Con la subsiguiente expresión transitoria a nivel local de la proteína S, ésta será reconocida como antígeno extraño por las células del sistema inmunitario –como neutrófilos y células presentadoras de antígenos (células dendríticas y macrófagos, fundamentalmente)–, que son reclutadas como consecuencia de la inflamación local transitoria producida por la inyección. Se desencadena, en última instancia, una respuesta adaptativa contra el antígeno tanto de anticuerpos neutralizantes y otros anticuerpos funcionales específicos anti-S (mediada por linfocitos B) como de inmunidad celular (mediada por linfocitos T, particularmente a través de una respuesta regulada por Th1), lo que puede contribuir a la protección contra la COVID-19 sintomática ante una posible exposición futura al SARS-CoV-2. En principio, los anticuerpos neutralizantes específicos generados serán capaces de prevenir la infección mediante el bloqueo de la interacción entre la proteína S y su dominio de unión al receptor (RBD, por sus siglas en inglés) con la célula del hospedador, pero también por aclaramiento viral mediante el proceso de opsonización.
Es una vacuna que ha sido específicamente diseñada para proporcionar a las células humanas la secuencia genética codificante para la proteína S del SARS-CoV-2, cuya función se ha definido en el apartado anterior. A diferencia de las estrategias “clásicas” para inducir respuesta inmunitaria empleadas por la mayoría de vacunas hasta ahora disponibles (por ejemplo, la inclusión de subunidades proteicas o de virus vivos atenuados/virus inactivados), basadas en usar antígenos del microorganismo, ésta contiene material genético del virus (fragmentos de su ADN), sin introducir el virus o sus partes en sí. O sea, al contener un vector de adenovirus sin capacidad replicativa y no contener SARS-CoV-2, la administración de la vacuna no puede producir enfermedad por adenovirus ni COVID-19. Cabe destacar que, a diferencia de la vacuna de AstraZeneca, la secuencia codificante de esta vacuna sí ha sido modificada para que se exprese una proteína S en conformación estabilizada, diferente a la proteína del SARS CoV 2 wild type (en la conformación trimérica prefusión), de forma similar a lo que se comentó para las dos vacunas de ARNm ya autorizadas (Fernández-Moriano, 2021a).
Diversos estudios bioquímicos in vitro y ensayos in vivo en modelos animales murinos, de hámsteres, de conejos, y de primates no humanos permitieron demostrar que la vacuna AD26.COV2-S es suficientemente inmunógena, esto es, induce una respuesta inmunitaria específica consistente en términos de producción de anticuerpos IgG y de respuesta de células T, que resultaba notablemente protectora frente a la exposición al virus (EMA, 2021).
Aspectos moleculares
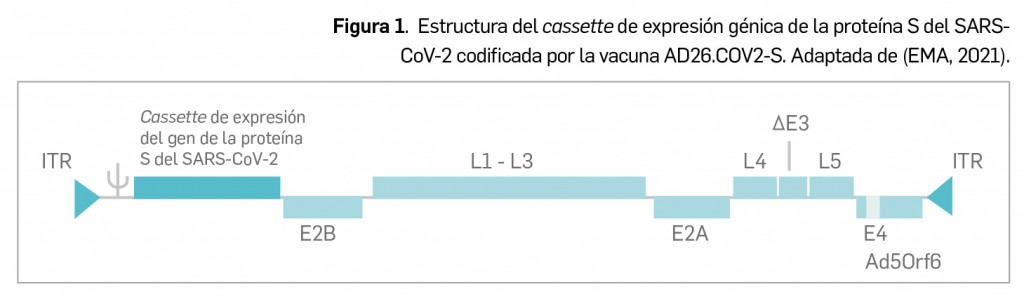
Se trata de una vacuna a base de adenovirus humano tipo 26 (Ad26)6, producido en células de la línea celular PER.C6 TetR (derivada de células retinianas embrionarias humanas) mediante tecnología de ADN recombinante, que ha sido modificado genéticamente para eliminar su capacidad de replicación por deleciones en el gen E1 (ΔE1A/E1B) y de una parte del gen E3 (ΔE3) –para generar suficiente espacio en el genoma viral donde insertar las regiones de antígenos externos– y la sustitución de la región Ad26 E4 orf6 por la homóloga del adenovirus tipo 5 –a fin de permitir la replicación del vector en líneas celulares–.
El vector adenoviral consiste en viriones no envueltos de entre 80-100 nm de diámetro, en los que la molécula de ADN linear de aproximadamente 35.000 pares de bases se encapsula en una estructura icosaédrica formada por siete proteínas estructurales, llamadas proteína II (hexon), III (penton), IV (fibre), VI, VIII, IX y IIIa, y cuatro proteínas centrales (V, VII, X y la proteína terminal) que están directamente asociadas con la molécula de ADN. El transgén contenido en la región ΔE1A/E1B del vector adenoviral (Figura 1) codifica para una proteína S de cadena completa, modificada para incluir mutaciones estabilizadoras en la conformación prefusión, tales como 2 cambios de aminoácidos en la zona de unión de las subunidades S1 y S2 (que eliminan el sitio de escisión por furina) y dos sustituciones de prolina en la región bisagra de la proteína.
Eficacia y seguridad clínicas
A pesar de la emergencia sanitaria que representa la COVID-19, la vacuna AD26.COV2-S ha sido –y continúa siendo– adecuadamente evaluada, conforme a los estándares regulatorios vigentes, en ensayos clínicos de fase 3 suficientemente amplios, con diseño adaptativo, multicéntrico y randomizado. Así, la autorización condicional en la Unión Europea se ha basado en los datos provisionales del estudio pivotal ENSEMBLE (COV3001), aún en marcha. Se trata de un ensayo aleatorizado (1:1), doble ciego y controlado por placebo desarrollado en 213 centros de 8 países (EE.UU., Sudáfrica y países de Latinoamérica, incluyendo Brasil) que ha enrolado a un total de 44.325 participantes adultos de 18 años de edad o mayores, quienes recibieron una dosis intramuscular de la vacuna (N= 21.895) o placebo (N= 21.888); no podían recibir ninguna otra vacuna en los 14 días previos y posteriores a la vacunación.
Se excluyeron pacientes con confirmación de seropositividad para el SARS-CoV-2, disfunción del sistema inmunitario (no pacientes con infección por VIH estabilizada por terapia antirretroviral), en tratamiento inmunosupresor reciente (< 6 meses) y mujeres embarazadas. Las características demográficas estuvieron bien equilibradas entre los dos brazos de tratamiento, destacando, entre los participantes que recibieron la vacuna, las siguientes: edad media de 52,0 años (80% tenía entre 18 y 64 años, el 20% tenía ≥ 65 años y, de éstos, el 4% tenía ≥ 75 años), el 44% eran mujeres, el 47% eran de EE.UU., el 41% de latinoamérica y el 13% de Sudáfrica. Además, el 40% de los individuos tenían al menos una comorbilidad asociada con un mayor riesgo de agravamiento de la COVID-19, fundamentalmente obesidad (28%), hipertensión (10%), diabetes tipo 2 (7%), infección por VIH (2,5%), enfermedades cardiacas graves (2,4%) y asma (1,3%).
Con una mediana de seguimiento de 58 días (rango 1-124 días) posvacunación, el análisis principal de eficacia se basó en los datos clínicos de 39.321 participantes, incluyendo 1.262 para quienes el estado serológico basal era desconocido. Los resultados (EMA, 2021; AEMPS, 2021) ponen de manifiesto que, a los 14 días tras la vacunación, la eficacia global de la vacuna en la prevención de casos sintomáticos de COVID-19 se situó en un 66,9% (IC95% 59,0-79,4), habida cuenta de la confirmación –mediante prueba PCR positiva y ≥ 1 signo/síntoma respiratorio o ≥ 2 signos/síntomas sistémicos– de 116 casos de COVID-19 en el grupo de la vacuna y 348 en el grupo control, con un seguimiento similar en ambos brazos (3.116 y 3.096 años-persona, respectivamente). Si se considera el número de casos de COVID-19 sintomática confirmados con inicio al menos 28 días después de la vacunación (que fue la otra variable co-primaria predefinida) –66 entre quienes recibieron la vacuna y 193 entre quienes recibieron placebo–, la eficacia vacunal fue pareja, alcanzando el 66,1% (IC95% 55,0-74,8), también con seguimientos similares entre grupos (3.102 y 3.070 años-persona).
De modo interesante, esa eficacia fue consistente entre los distintos subgrupos etarios, alcanzando valores de inmunoprevención superiores en los participantes de mayor edad. Así, por ejemplo, a los 14 días posvacunación se estimó una eficacia del 64,2% entre 18 y 64 años (IC95% 55,2-71,6; 107 casos con la vacuna vs. 297 con placebo), que ascendía al 82,4% en los participantes de ≥ 65 años (IC95% 63,9-92,4; 9 casos con la vacuna vs. 51 con placebo) y hasta el 100% en sujetos de ≥ 75 años (IC95% 45,9-100; 0 casos con la vacuna vs. 8 con placebo). Si se consideran los casos de COVID-19 de inicio ≥ 28 días tras la vacunación, esas tasas de eficacia son similares: 65,1% entre los 18 y 64 años (IC95% 52,9-74,5; 60 casos con la vacuna vs. 170 con placebo) y 74,0% en participantes de ≥ 65 años (IC95% 34,4-91,4; 6 casos con la vacuna vs. 23 con placebo). Los análisis por subgrupos también revelaron que la eficacia es independiente del sexo o de la presencia o ausencia de comorbilidades médicas típicamente asociadas a una mayor severidad de la patología.
Además, no menos importante, la eficacia de la vacuna en la prevención de casos graves de la patología –definidos, como mínimo, por el criterio de SpO2 ≤ 93% en aire ambiente– en adultos seronegativos fue del 76,7% (IC95% 54,6-89,1; 14 casos con la vacuna vs. 60 con placebo) en los 14 días tras la vacunación, y del 85,4% (IC95% 54,2-96,9; 5 casos con la vacuna vs. 34 con placebo) si se consideraban solo los casos de inicio de síntomas ≥ 28 días tras la vacunación. De entre los casos graves con inicio al menos 14 días después de la vacunación, se produjeron 8 hospitalizaciones, solo 2 de las cuales ocurrieron entre los sujetos vacunados; y hubo 6 muertes por COVID-19, todas ellas en el grupo placebo. Más allá de los 28 días, se registraron 10 hospitalizaciones por la enfermedad, solo 2 entre los sujetos vacunados; 1 de los casos del grupo placebo requirió ingreso en UCI y ventilación mecánica. Tales hallazgos fueron respaldados por un análisis post-hoc de todas las hospitalizaciones relacionadas con COVID-19, en una búsqueda más amplia en toda la información disponible de cualquier fuente (2 vs. 29 casos en el conjunto de datos extendido).
Finalmente, se realizó un análisis exploratorio de eficacia con los subgrupos de participantes en Brasil, Sudáfrica y Estados Unidos, a fin de evaluar la posible influencia de las distintas variables virales: con una secuenciación de casi el 72% de los casos confirmados por laboratorio central (aunque con cierto desequilibrio en las muestras secuenciadas entre los brazos del estudio), en EE.UU. se identificaron un 96% de las muestras como la variante “clásica” D614G (Wuhan-H1), en Sudáfrica el 95% de las cepas fueron de la variante B.1.351 (también llamada 20H/501Y.V2), y en Brasil el 70% de las cepas fueron de la variante P.2, correspondiendo el 30% restante a la variante Wuhan-H1. Si se consideran los datos de infecciones confirmadas pasados los 28 días de recibir la dosis, la eficacia vacunal varió ligeramente según el área geográfica, de forma que llegó al 72,0% en Estados Unidos, al 68,1% en Brasil, y se mantenía en el 64,0% en Sudáfrica. Se estimó que, para evitar los casos de enfermedad grave/crítica, la eficacia de la vacuna es mayor, moviéndose en esos tres países en el rango del 82-88% (85,9% en EE.UU., 87,6% en Brasil y 81,7% en Sudáfrica).
En cuanto a su seguridad, los datos más sólidos proceden del estudio pivotal, donde un total de 21.895 adultos recibieron la vacuna, la cual parece bien tolerada. El análisis se realizó cuando se disponía de una mediana de seguimiento de más de 2 meses en casi 12.000 sujetos, y puso de manifiesto que la mayoría de los eventos adversos se notificaron en el plazo de 1-2 días después de la vacunación y fueron de intensidad leve o moderada y autolimitados en duración (1-2 días). La reacción local notificada con mayor frecuencia fue el dolor en el lugar de inyección (48,6%) y, entre las reacciones adversas sistémicas, destacaron: cefalea (38,9%), fatiga (38,2%), mialgia (33,2%), náuseas (14,2%) y fiebre (9%). La reactogenicidad de la vacuna fue más leve y menos frecuente en personas de ≥ 65 años (AEMPS, 2021), y se puede manejar fácilmente mediante un tratamiento sintomático con analgésicos o antipiréticos (como paracetamol7). En resumen, en los ensayos clínicos se han observado menos efectos secundarios que con otros fármacos (solo 5 voluntarios tuvieron eventos secundarios graves: 2 parálisis facial, 1 síndrome Guillain-Barré, 1 hipersensibilidad y 1 pericarditis) y ninguna reacción alérgica, constituyendo un perfil de seguridad similar al de otras vacunas y consistente, sin variaciones en función de la raza, las comorbilidades o la evidencia previa de infección por SARS-CoV-2 antes de la vacunación.
Cabe destacar que, habiéndose vacunado con este medicamento más de 7 millones de personas en EE.UU., a fecha de 13 de abril de 2021, cuando no se había iniciado aún su distribución en la UE, las autoridades sanitarias de aquel país recomendaron detener temporalmente la administración de esta vacuna, como precaución mientras evalúan la señal de seguridad surgida con la notificación de 8 eventos tromboembólicos en sujetos vacunados8 (supondrían el 0,0008% de las personas inmunizadas). Se trata de casos, con una frecuencia extremadamente rara, de coágulos sanguíneos atípicos y graves denominados trombosis de seno venoso cerebral, que se asocian a trombocitopenia. Los casos por ahora notificados han ocurrido entre mujeres de 18-48 años, y sus síntomas se han manifestado entre 6 y 13 días tras la vacunación. Es una señal de seguridad similar a la descrita para Vaxzevria®, la vacuna de AstraZeneca, que fue comentada ampliamente en el anterior número de Panorama Actual del Medicamento.
Aspectos innovadores
La nueva vacuna monovalente AD26.COV2-S es un fármaco compuesto por un vector viral único de adenovirus tipo 26 humano (Ad26) no replicativo que contiene un ADN codificante para la glicoproteína S del SARS CoV 2 de longitud completa y en una conformación prefusión estabilizada, para lo que el ADN se modifica con mutaciones específicas. La expresión transitoria de la proteína S a nivel local por las células huésped en la zona de inyección (tras la transcripción del ADN a ARNm en el núcleo celular y la posterior traducción a la proteína en el ribosoma citoplasmático) permitirá que sea reconocida como antígeno extraño por las células del sistema inmunitario, desencadenando una respuesta adaptativa tanto de anticuerpos neutralizantes como de inmunidad celular por linfocitos T, que contribuirán a la protección frente a COVID‑19 sintomática ante una futura exposición al SARS-CoV-2. En base a ello, el medicamento ha sido autorizado para la inmunización activa para prevenir la COVID-19 en personas de 18 años de edad y mayores. Se administra por vía intramuscular en una pauta de dosis única (≥ 5×1010 partículas virales, que se corresponden con 8,92 log10 unidades infecciosas en 0,5 ml), debiéndose usar siempre siguiendo las líneas de la “Estrategia de vacunación frente a COVID-19 en España” del Ministerio de Sanidad9.
A pesar de la emergencia de salud pública que representa la pandemia por COVID-19, la nueva vacuna ha sido adecuadamente evaluada en su indicación y dosis, conforme a los estándares de calidad vigentes en investigación clínica, en un ensayo pivotal controlado de fase 3 considerado aceptable por la EMA (por su diseño aleatorizado, ciego y multicéntrico, y por la amplia muestra de participantes). En base a los datos generados, le ha otorgado una autorización condicional de comercialización, supeditada a la obtención de nuevos datos clínicos en un futuro, que serán revisados al menos una vez al año, y que ha sido alcanzada en plazos excepcionalmente acortados gracias a la implementación de procesos de evaluación continua (rolling review) de la evidencia científica, que garantizan el cumplimiento de los requisitos de seguridad, eficacia y calidad.
Dicha autorización se ha sustentado fundamentalmente en los resultados del análisis principal de eficacia del citado estudio, con datos que derivan de más de 44.000 participantes adultos en EE.UU., Latinoamérica y Sudáfrica, de los cuales casi 21.900 recibieron la vacuna (media de edad de 52 años); un 40% de ellos presentaba alguna comorbilidad asociada a un mayor riesgo de agravamiento de la COVID-19 (28% de obesos). Con un seguimiento de unos dos meses, la administración de la vacuna en los sujetos seronegativos para SARS-CoV-2 demostró una eficacia protectora del 66,9% en la prevención de casos sintomáticos de la enfermedad de inicio ≥ 14 días desde la vacunación (116 casos confirmados en el grupo de la vacuna vs. 348 en el grupo placebo), y del 66,1% pasados 28 días (66 vs. 193 casos confirmados). La eficacia de la vacuna se mostró independiente de factores como el sexo, la presencia de comorbilidades, e incluso de la edad, aportando una inmunoprotección superior en sujetos de mayor edad: respecto a los casos de inicio tras ≥ 14 días, fue del 64% entre 18 y 64 años y de hasta el 82% en los participantes de ≥ 65 años; después de ≥ 28 días desde la vacunación, esas tasas de eficacia fueron del 65% y del 74%, respectivamente. La información en sujetos de > 75 años es limitada por la escasa población incluida (< 5%).
De modo interesante, la vacunación previno notablemente los casos graves de COVID-19, con tasas de eficacia del 76,7% (14 casos con la vacuna vs. 60 con placebo) tras 14 días y del 85,4% (5 casos con la vacuna vs. 34 con placebo) si se consideran los casos de inicio de síntomas después de ≥ 28 días. Así, más allá de las 4 semanas posvacunación, se produjeron 10 hospitalizaciones por la enfermedad, solo 2 entre los sujetos vacunados, y todas las muertes por COVID-19 registradas en el estudio (6) ocurrieron en el grupo placebo. Adicionalmente, un análisis exploratorio en las distintas regiones geográficas aportó una visión de la influencia de las variables virales: la eficacia de la vacuna, aunque ligeramente menor, parece mantenerse en niveles de protección aceptables frente a la variante B.1.351 (“surafricana”) y la variante P.2 (“brasileña”). Así, hubo solo pequeñas variaciones entre EE.UU. (donde la prevención de COVID-19 sintomática tras ≥ 28 días desde la vacunación fue del 72%), Brasil (68%) y Sudáfrica (64%); para la prevención de casos graves/críticos las diferencias fueron menores, manteniendo una eficacia importante en todas las regiones y ante todas las variantes virales (86% en EE.UU., 88% en Brasil y 82% en Sudáfrica).
Como se comentaba para las tres vacunas previamente autorizadas (Fernández-Moriano, 2021a; Fernández-Moriano, 2021b), persisten una serie de limitaciones relativas a la eficacia vacunal. Además de la ya citada en personas de más de 75 años, en este caso las incertidumbres se refieren fundamentalmente a la prevención frente al ingreso en UCI o frente a la mortalidad (por el escaso número de eventos registrados), o frente a los casos de COVID-19 permanente. Tampoco se ha concluido sobre la eficacia frente a la infección asintomática y la capacidad de transmitir la enfermedad, ni sobre el tiempo de la inmunidad conferida por la vacuna. También hay dudas sobre la protección frente las nuevas variantes virales, para lo que se requieren estudios confirmatorios.
En términos de seguridad, esta nueva vacuna parece bien tolerada a corto plazo, con un perfil toxicológico similar al de otras vacunas autorizadas y consistente entre subgrupos de pacientes con independencia de la raza, comorbilidades o evidencia de infección previa por el coronavirus. La mayoría de eventos adversos se registraron a corto plazo tras la vacunación (1-2 días), fueron leves-moderados en intensidad y autolimitados en duración (1-2 días). Destacan por su frecuencia los siguientes: dolor en el lugar de inyección (49%), cefalea (39%), fatiga (38%), mialgia (33%) y náuseas (14%); la reactogenicidad de la vacuna, clínicamente manejable con facilidad, se reveló menor en personas mayores de 65 años. Es preciso recordar que el perfil de eventos adversos es desconocido a medio-largo plazo (más allá de los 2 meses), y persiste el riesgo de que no en los estudios clínicos no se hayan detectado eventos adversos poco frecuentes (< 1 caso cada 10.000). Habrá que esperar a los datos de seguridad tras el seguimiento de 2 año previsto en el ensayo pivotal, no pudiéndose descartar aún la posibilidad de anafilaxia ni de aparición de eventos trombóticos raros.
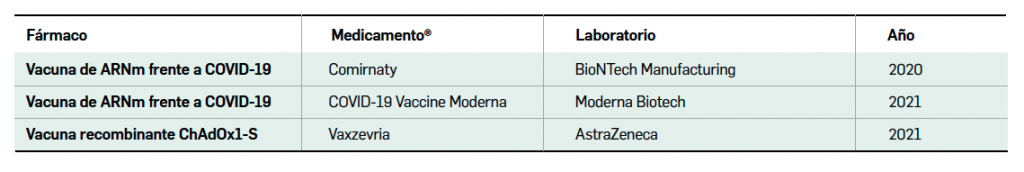
En definitiva, dado el contexto vigente de alta transmisión comunitaria de la COVID-19, la emergencia de nuevas variantes virales y la necesidad global de disponer de suficientes medicamentos profilácticos autorizados, esta nueva vacuna es la primera que se autoriza en pauta de 1 dosis, por lo que representa cierto grado de avance terapéutico en su indicación, sin suponer una innovación farmacológica disruptiva, pues el uso de vectores adenovirales en vacunas de uso humano no es una estrategia tan novedosa como lo fue el ARNm (ya se dispone de otras vacunas a base de vectores virales incluso frente a la COVID-19, como es el caso de Vaxzevria®). Con un perfil de seguridad similar al resto de vacunas aprobadas en la UE, los datos clínicos apuntan a una inmunoprotección frente a la enfermedad sintomática inferior a la que aportan las vacunas de ARNm BNT162b2 (Comirnaty®) y mRNA-1273 (COVID-19 Vaccine Moderna®), las cuales se mueven en tasas de eficacia del 94-95%, pero similar o superior a ChAdOx1-S (Vaxzevria®), cercana al 60%. No obstante, el grado de prevención de la COVID-19 grave (> 85%) es muy relevante clínicamente. Contribuye a ampliar el arsenal terapéutico para elevar las coberturas vacunales en un contexto de emergencia sanitaria mundial, aportando otra ventaja interesante, en comparación con las vacunas de ARNm, que se refiere a su mayor estabilidad: no requiere necesariamente congelación y se puede conservar hasta 3 meses en nevera (2-8ºC) e incluso 12 horas a temperatura ambiente.
Valoración

Fármacos relacionados registrados en España