Resumen
El Consejo General de Colegios de Farmacéuticos, a través de su base de datos BOT PLUS, permite a los profesionales sanitarios conocer la información relativa a las condiciones de empleo de los medicamentos durante la lactancia, en base a la información oficial disponible en las fichas técnicas. Se resume a continuación la información a la que se puede acceder y la manera de encontrar esta información en BOT PLUS.
Durante la lactancia natural, el recién nacido puede sufrir los efectos de los medicamentos con los que se trate la madre, debido a su posible eliminación por la leche materna. Existen múltiples factores, dependientes de la madre, del niño y del propio medicamento, que intervienen en la capacidad que tiene un medicamento para alcanzar la leche y ser capaz de ejercer su acción en el niño que está siendo amamantado. Entre estos factores se encuentran: las propiedades físicas y químicas del medicamento, el flujo sanguíneo mamario (debiéndose prestar atención, por tanto, a los medicamentos o circunstancias que modifiquen el flujo sanguíneo), el control hormonal, la posología con que se administra el medicamento a la madre, el reflejo succionador del niño, y la cantidad y periodicidad de las tomas.
En principio, no hay que recomendar la interrupción de la lactancia, puesto que la mayoría de los medicamentos no comportan ningún riesgo para la salud del niño. Además, el hecho de que un medicamento se excrete en la leche materna no implica necesariamente toxicidad, ya que tendría que alcanzar determinadas concentraciones para originar efectos adversos en el lactante y, en la mayoría de los casos, los niveles plasmáticos alcanzados son de escasa relevancia clínica.
En cualquier caso, es necesario que un profesional sanitario evalúe individualmente la relación beneficio para la madre/riesgo para el niño. Para ayudar al farmacéutico con este análisis, en BOT PLUS está disponible la información sobre el posible uso de los medicamentos durante la lactancia materna, procedente de una fuente oficial como es la ficha técnica del medicamento. Si se decide la administración del medicamento y la continuación de la lactancia materna, se deberá observar estrechamente al niño ante cualquier posible manifestación de efectos tóxicos (sedación, trastornos en el crecimiento, etc.).
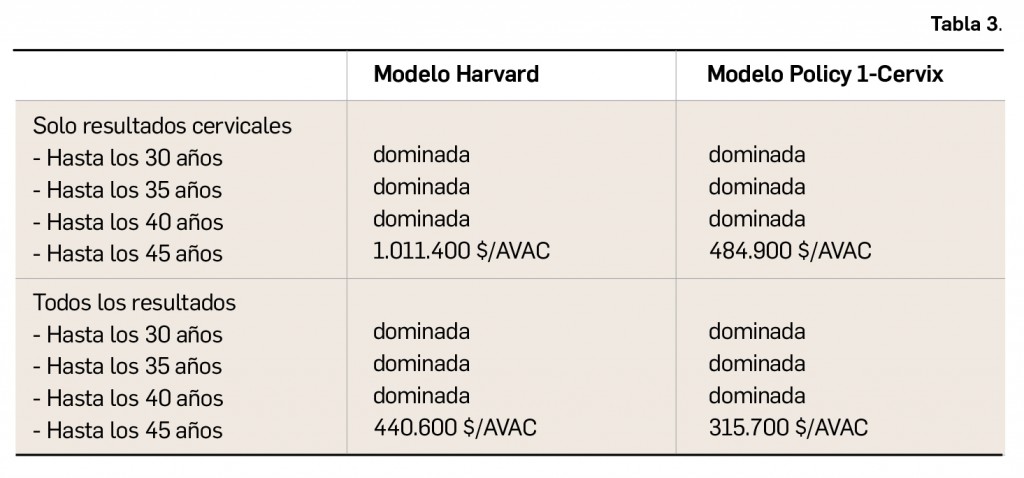
Para ayudar al farmacéutico en su labor asistencial, en la base de datos BOT PLUS se han clasificado los medicamentos en tres grupos en base a su posible uso durante la lactancia, asignándoles un mensaje de advertencia específico, así como un pictograma con el que se puede comprobar de forma rápida en la ficha de cada medicamento si éste se puede administrar en mujeres lactantes o no (Tabla 1). Toda la información relativa al uso de los medicamentos durante la lactancia procede de las fichas técnicas autorizadas por la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) para cada medicamento y, tras su análisis, se codifica en BOT PLUS.

simismo, podemos encontrar mensajes de advertencia secundarios relacionados con la lactancia que advertirían de consideraciones especiales a tener en cuenta, como, por ejemplo, durante cuánto tiempo se debe suspender la lactancia materna tras la administración de un medicamento, o contraindicaciones específicas.
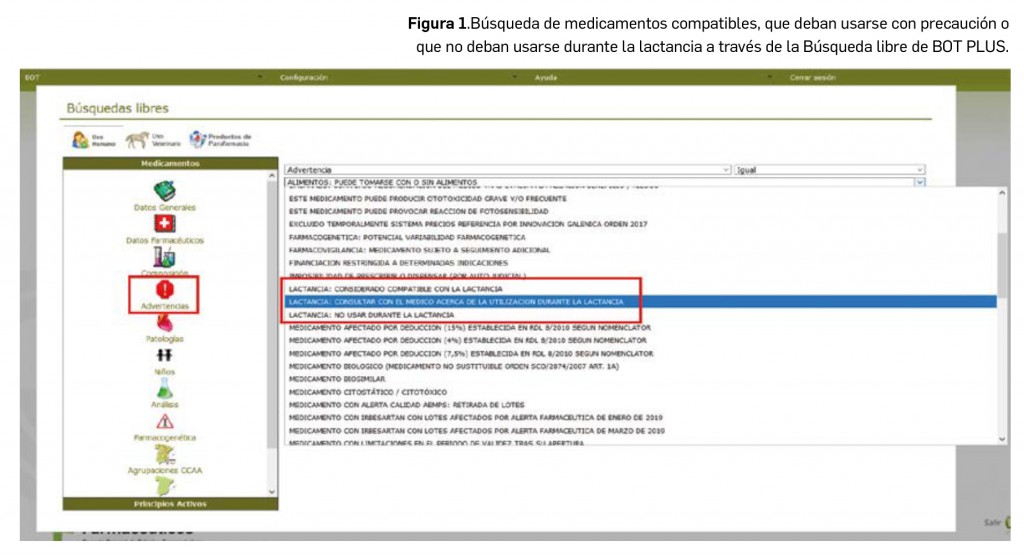
En el caso de querer obtener un listado de todos aquellos medicamentos que sean compatibles, deban usarse con precaución o no deban usarse durante la lactancia, podemos realizar una búsqueda libre en BOT PLUS, pinchando para ello el botón de “Búsquedas libres” en la pantalla de inicio de la aplicación. Dentro del buscador, iremos añadiendo las condiciones necesarias para obtener el listado deseado. En este caso específico, dentro del icono Advertencias encontraremos los mensajes relacionados con la lactancia (Figura 1).
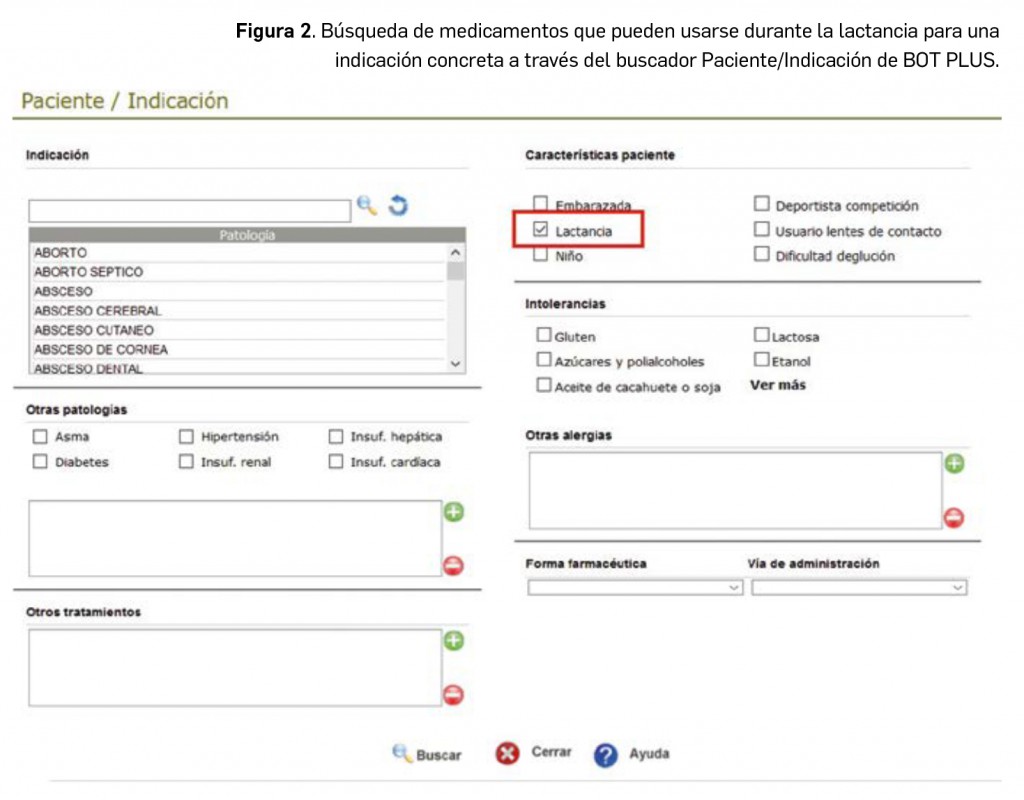
dicionalmente, BOT PLUS permite obtener un listado de medicamentos para una indicación concreta en un paciente concreto, mediante la funcionalidad del Buscador Paciente/Indicación. En este buscador puede introducirse tanto la indicación para la cual se está realizando la búsqueda como otras patologías, tratamientos, características del paciente, intolerancias, alergias y excipientes a evitar. En este caso, en el apartado de características del paciente seleccionaremos “lactancia” y, así, todos los medicamentos que se obtengan en la búsqueda para la indicación concreta serán aptos (no aparecerán en el listado los medicamentos que no deben usarse) en estas pacientes mujeres en periodo de lactancia (Figura 2), si bien pudiera ser que debieran usarse exclusivamente bajo recomendación del médico.
En definitiva, mediante la codificación de esta información sobre el uso de los medicamentos autorizados por la AEMPS durante la lactancia materna mediante la inclusión de mensajes de advertencia en BOT PLUS, el farmacéutico comunitario puede identificar fácilmente aquellos medicamentos que pueden utilizarse en mujeres lactantes o no, facilitando así su labor asistencial.

 El primero, Francisco Javier Balmis, dio nombre a la operación desplegada por la Unidad Militar de Emergencias en favor de los infectados, con una eficacia espléndida, luego olvidada en aras de las disputas políticas y gremiales.
El primero, Francisco Javier Balmis, dio nombre a la operación desplegada por la Unidad Militar de Emergencias en favor de los infectados, con una eficacia espléndida, luego olvidada en aras de las disputas políticas y gremiales. El año 1799, el médico italiano Luigi Careno (1766-1810) tradujo al latín el texto de Jenner
El año 1799, el médico italiano Luigi Careno (1766-1810) tradujo al latín el texto de Jenner La vacunación se extendió por toda España en los primeros años del siglo XIX. Las iniciales discusiones científicas giraron en torno a dos cuestiones. En primer lugar, la eficacia de la misma, por tratarse, en principio, de la inoculación de una zoonosis (enfermedad que afectaba solo a los animales y no a los humanos). Los resultados empíricos demostraron su utilidad. En segundo lugar, temían el efecto de la vacuna: desconocían si con ella sucedería lo mismo que con la variolización, o inoculación de linfa procedente de personas con la viruela humana, y pudiera potenciarse la enfermedad en lugar de prevenirla. Los resultados prácticos demostraron también lo inadecuado del temor.
La vacunación se extendió por toda España en los primeros años del siglo XIX. Las iniciales discusiones científicas giraron en torno a dos cuestiones. En primer lugar, la eficacia de la misma, por tratarse, en principio, de la inoculación de una zoonosis (enfermedad que afectaba solo a los animales y no a los humanos). Los resultados empíricos demostraron su utilidad. En segundo lugar, temían el efecto de la vacuna: desconocían si con ella sucedería lo mismo que con la variolización, o inoculación de linfa procedente de personas con la viruela humana, y pudiera potenciarse la enfermedad en lugar de prevenirla. Los resultados prácticos demostraron también lo inadecuado del temor.
 El alicantino Francisco Javier Balmis Berenguer, en su calidad de cirujano militar, había participado en el sitio de Gibraltar (1780) y servido once años en la América hispana, en las Antillas y México. Cursó estudios de Medicina, recibió el título de doctor y en 1795, de regreso en España, fue nombrado cirujano honorario de Cámara de Carlos IV, además de tratar de introducir varios elementos de la terapéutica americana en Europa.
El alicantino Francisco Javier Balmis Berenguer, en su calidad de cirujano militar, había participado en el sitio de Gibraltar (1780) y servido once años en la América hispana, en las Antillas y México. Cursó estudios de Medicina, recibió el título de doctor y en 1795, de regreso en España, fue nombrado cirujano honorario de Cámara de Carlos IV, además de tratar de introducir varios elementos de la terapéutica americana en Europa. El suero vacunal se llevó desde Madrid a la Coruña, brazo a brazo, mediante cinco niños de la Casa de Desamparados de la Corte, uno de los cuales murió durante el viaje. El fluido podía llevarse también entre cristales, pero el método era poco fiable y solo servía para recorridos muy cortos. En ese momento, se consideraba mejor proceder a vacunar con el líquido procedente de las pústulas de vacas infectadas a un niño. Cuando desarrollaba sus propias postillas se vacunaba a otro, con el pus procedente de ellas, y así se podía trasportar indefinidamente, si el número de receptores era el adecuado. Se preferían los niños porque se sabía ya que, al vacunar brazo a brazo entre adultos, se podían transmitir otras enfermedades.
El suero vacunal se llevó desde Madrid a la Coruña, brazo a brazo, mediante cinco niños de la Casa de Desamparados de la Corte, uno de los cuales murió durante el viaje. El fluido podía llevarse también entre cristales, pero el método era poco fiable y solo servía para recorridos muy cortos. En ese momento, se consideraba mejor proceder a vacunar con el líquido procedente de las pústulas de vacas infectadas a un niño. Cuando desarrollaba sus propias postillas se vacunaba a otro, con el pus procedente de ellas, y así se podía trasportar indefinidamente, si el número de receptores era el adecuado. Se preferían los niños porque se sabía ya que, al vacunar brazo a brazo entre adultos, se podían transmitir otras enfermedades. El 9 de diciembre de 1803, fondearon en Santa Cruz de Tenerife a las ocho de la noche. Una hora después ya estaban vacunados diez niños de las principales familias de la isla, con pus extraído de los cuatro que habían desembarcado con Balmis. La recepción de las autoridades civiles, militares y eclesiásticas fue entusiástica y agradecida, aunque se temió que la expedición se llevase niños de las islas, lo que fue rápidamente desmentido.
El 9 de diciembre de 1803, fondearon en Santa Cruz de Tenerife a las ocho de la noche. Una hora después ya estaban vacunados diez niños de las principales familias de la isla, con pus extraído de los cuatro que habían desembarcado con Balmis. La recepción de las autoridades civiles, militares y eclesiásticas fue entusiástica y agradecida, aunque se temió que la expedición se llevase niños de las islas, lo que fue rápidamente desmentido. Balmis pasó a La Habana, desde La Guaira, a bordo del María Pita. Partió el 8 de mayo y llegó el 26 del mismo mes. También allí, como en San Juan de Puerto Rico, el doctor Tomás Romay Chacón
Balmis pasó a La Habana, desde La Guaira, a bordo del María Pita. Partió el 8 de mayo y llegó el 26 del mismo mes. También allí, como en San Juan de Puerto Rico, el doctor Tomás Romay Chacón Salvany comenzó a manifestar síntomas de una tuberculosis pulmonar con vómitos de sangre, pero, enterado de una grave incidencia de la viruela en la Audiencia de Quito (Ecuador), salió hacia allí. Llegó el 16 de julio de 1806, junto a Rafael Lozano y cuatro niños. Vacunó a unas siete mil personas. Pasó a Loja (Ecuador) y cayó gravemente enfermo en Piura (Perú).
Salvany comenzó a manifestar síntomas de una tuberculosis pulmonar con vómitos de sangre, pero, enterado de una grave incidencia de la viruela en la Audiencia de Quito (Ecuador), salió hacia allí. Llegó el 16 de julio de 1806, junto a Rafael Lozano y cuatro niños. Vacunó a unas siete mil personas. Pasó a Loja (Ecuador) y cayó gravemente enfermo en Piura (Perú). El 3 de septiembre de 1805 Balmis embarcó, en Manila, con Francisco Pastor Balmis y tres niños, en una nave portuguesa, La Diligencia, que le llevó a Macao (China); la travesía fue muy dura, pues les sorprendió un tifón. Recibió la ayuda del Gobernador portugués y del obispo y fueron muchos los vacunados. Se internó en China, llegando a Cantón el 5 de octubre de 1805, donde vacunó a muy pocas personas, pues no consiguió la cooperación de los españoles, delegados de la Compañía de Filipinas en aquella ciudad, con la que se tenían negocios (Figura 10).
El 3 de septiembre de 1805 Balmis embarcó, en Manila, con Francisco Pastor Balmis y tres niños, en una nave portuguesa, La Diligencia, que le llevó a Macao (China); la travesía fue muy dura, pues les sorprendió un tifón. Recibió la ayuda del Gobernador portugués y del obispo y fueron muchos los vacunados. Se internó en China, llegando a Cantón el 5 de octubre de 1805, donde vacunó a muy pocas personas, pues no consiguió la cooperación de los españoles, delegados de la Compañía de Filipinas en aquella ciudad, con la que se tenían negocios (Figura 10).