La evolución de la pandemia y la amplia capacidad diagnóstica de algunos países han permitido el desarrollo de estudios a nivel poblacional que permiten ampliar el conocimiento sobre la epidemiología de la COVID-19. Como ejemplo se puede poner un trabajo desarrollado en Dinamarca (país donde las pruebas diagnósticas PCR son gratuitas y pueden acceder a ellas tanto ciudadanos sintomáticos como asintomáticos), el más amplio sobre el riesgo de reinfección por SARS-CoV-2 realizado hasta la fecha. Se habían publicado previamente varios estudios, más pequeños y limitados, que apuntaban a tasas de reinfección menores al 1%. Esto se confirma ahora con una evidencia más robusta.
Los investigadores realizaron un estudio observacional retrospectivo con datos de aproximadamente 4 millones de individuos (casi el 70% de la población danesa) que durante 2020 habían sido sometidos a 10,6 millones de pruebas PCR, y analizaron la incidencia de infecciones durante la segunda ola de la epidemia en aquel país (entre los meses de septiembre y diciembre de 2020) para compararla con la proporción de ciudadanos con resultados PCR positivos y negativos durante la primera ola (de marzo a mayo de ese año); del análisis principal se excluyeron datos de personas con primoinfección entre ambos periodos y de fallecidos antes de la segunda ola. Adicionalmente, un análisis de cohortes alternativo comparó las tasas de infección durante el año entre ciudadanos con y sin confirmación de la infección en los 3 meses previos, investigando posibles diferencias por edad, sexo o tiempo desde la infección.
Durante la primera ola se evaluó a 533.381 personas, de las cuales 11.727 (2,2%) tuvieron una PCR positiva; una gran proporción de ellos fueron elegibles para el seguimiento durante la segunda ola (525.339), de los cuales 11.068 (2,11%) habían sido positivos en la primera. Entre estos últimos, solo 72 pacientes (0,65%; IC95% 0,51-0,82) tuvieron un nuevo resultado positivo durante la segunda ola en comparación con 16.819 positivos (3,27% de un total de 514.271 sujetos; IC95% 3,22-3,32) que no se habían infectado –tuvieron PCR negativa– durante la primera ola (RR ajustada= 0,195; IC95% 0,155-0,246). La protección contra la reinfección era del 80,5% (IC95% 75,4-84,5) entre quienes habían tenido una PCR positiva en la primera ola. No se detectó ningún caso de triple infección. El análisis alternativo de cohortes arrojó unas estimaciones similares (RR ajustado= 0,212 y protección frente a la reinfección del 78,8%), si bien reveló que aquellos individuos de 65 años o mayores presentaban una protección frente a la reinfección más reducida, del 47,1% (IC95% 24,7-62,8). No se observaron diferencias significativas, en cambio, en la protección frente a la reinfección en función del sexo (78,4% en hombres y 79,1% en mujeres) ni evidencia de una reducción de la protección según el tiempo desde la primoinfección, mantenida en torno al 80% al menos 6 meses (79,3% tras 3-6 meses de seguimiento vs. 77,7% tras ≥ 7 meses).
Estos hallazgos pueden contribuir a la priorización de determinados grupos poblacionales en la vacunación, incluyendo individuos previamente infectados, ya que prueban que la inmunidad “natural” por haber superado la infección no es del todo protectora, especialmente en personas mayores, que tienen un mayor riesgo de volver a contagiarse. Además, confirman que las reinfecciones son posibles, pero poco frecuentes, no jugando un papel demasiado relevante en la epidemiología de la enfermedad.
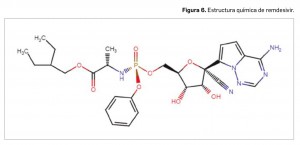 Fue el primer fármaco en ser autorizado frente a la COVID-19 en la Unión Europea. El 25 de junio de 2020 la Agencia Europea de Medicamentos (EMA) recomendó su autorización condicional
Fue el primer fármaco en ser autorizado frente a la COVID-19 en la Unión Europea. El 25 de junio de 2020 la Agencia Europea de Medicamentos (EMA) recomendó su autorización condicional Dexametasona (Figura 7) es un glucocorticoide fluorado de uso común desde la década de 1960 para tratar una amplia gama de afecciones con componente inflamatorio y autoinmune –entre ellas, artritis reumatoide, asma o procesos alérgicos o neoplásicos–, por sus propiedades antiinflamatorias, inmunosupresoras y antialérgicas, y su casi nula actividad mineralocorticoide. Está disponible en España en numerosas presentaciones de medicamentos para diversas vías de administración, siendo su perfil de seguridad no desdeñable, pero bien conocido (caracterizado por frecuentes eventos adversos tales como: susceptibilidad aumentada a las infecciones, candidiasis orofaríngea, hiperglucemia, osteoporosis, cataratas, insuficiencia adrenocortical e inducción de síntomas parecidos al síndrome de Cushing, etc.). La indicación autorizada para su uso en “COVID-19 en pacientes de ≥ 12 años y ≥ 40 kg que requieran oxígeno suplementario” ha sido incluido en la ficha técnica de varios medicamentos. No se ha probado, sin embargo, su beneficio en aquellos pacientes con enfermedad leve-moderada (una mayoría).
Dexametasona (Figura 7) es un glucocorticoide fluorado de uso común desde la década de 1960 para tratar una amplia gama de afecciones con componente inflamatorio y autoinmune –entre ellas, artritis reumatoide, asma o procesos alérgicos o neoplásicos–, por sus propiedades antiinflamatorias, inmunosupresoras y antialérgicas, y su casi nula actividad mineralocorticoide. Está disponible en España en numerosas presentaciones de medicamentos para diversas vías de administración, siendo su perfil de seguridad no desdeñable, pero bien conocido (caracterizado por frecuentes eventos adversos tales como: susceptibilidad aumentada a las infecciones, candidiasis orofaríngea, hiperglucemia, osteoporosis, cataratas, insuficiencia adrenocortical e inducción de síntomas parecidos al síndrome de Cushing, etc.). La indicación autorizada para su uso en “COVID-19 en pacientes de ≥ 12 años y ≥ 40 kg que requieran oxígeno suplementario” ha sido incluido en la ficha técnica de varios medicamentos. No se ha probado, sin embargo, su beneficio en aquellos pacientes con enfermedad leve-moderada (una mayoría).
 El Instituto de Salud de Montreal (Canadá) anunció los resultados del ensayo clínico de fase 3 COLCORONA (Tardif et al., 2021), controlado, doble ciego, multicéntrico y multinacional, que aleatorizó un total de 4.488 pacientes no hospitalizados con COVID-19 –confirmación por PCR o criterios clínicos– a recibir colchicina (0,5 mg/12 h durante 3 días, y posteriormente una vez al día) o placebo durante 30 días. La variable primaria de eficacia –una medida compuesta de muerte u hospitalización por COVID-19– se verificó en el 4,7% de los pacientes tratados con colchicina y en el 5,8% de los pacientes en el grupo placebo (OR= 0,79; IC95% 0,61-1,03; p= 0,08). Si se consideran solo los 4.159 pacientes con confirmación de la infección por PCR, la variable primaria ocurrió en el 4,6% de los tratados con colchicina frente al 6,0% de los pacientes en el grupo control (OR= 0,75; IC95% 0,57-0,99; p= 0,04). Concretamente, para ese grupo los autores refieren una reducción del 25% en las probabilidades de hospitalización (OR= 0,75; IC95% 0,57-0,99), del 50% en el riesgo de ventilación mecánica (OR= 0,50; IC95% 0,23-1,07) y del 44% en el riesgo de muerte (OR= 0,56; IC95% 0,19-1,66). Respecto al perfil toxicológico, se reportó una frecuencia de eventos adversos significativamente menor en el grupo de colchicina que en el grupo placebo (4,9% vs. 6,3%; p= 0,05), notificándose con menor frecuencia, por ejemplo, casos de neumonía (2,9% vs. 4,1% en el grupo control), si bien la diarrea fue más común en el grupo del fármaco (13,7% vs. 7,3%; p< 0,0001). Se debe recordar, no obstante, que la colchicina tiene un margen terapéutico estrecho y es extremadamente tóxica en caso de sobredosis, lo que requiere un ajuste muy riguroso de su pauta posológica.
El Instituto de Salud de Montreal (Canadá) anunció los resultados del ensayo clínico de fase 3 COLCORONA (Tardif et al., 2021), controlado, doble ciego, multicéntrico y multinacional, que aleatorizó un total de 4.488 pacientes no hospitalizados con COVID-19 –confirmación por PCR o criterios clínicos– a recibir colchicina (0,5 mg/12 h durante 3 días, y posteriormente una vez al día) o placebo durante 30 días. La variable primaria de eficacia –una medida compuesta de muerte u hospitalización por COVID-19– se verificó en el 4,7% de los pacientes tratados con colchicina y en el 5,8% de los pacientes en el grupo placebo (OR= 0,79; IC95% 0,61-1,03; p= 0,08). Si se consideran solo los 4.159 pacientes con confirmación de la infección por PCR, la variable primaria ocurrió en el 4,6% de los tratados con colchicina frente al 6,0% de los pacientes en el grupo control (OR= 0,75; IC95% 0,57-0,99; p= 0,04). Concretamente, para ese grupo los autores refieren una reducción del 25% en las probabilidades de hospitalización (OR= 0,75; IC95% 0,57-0,99), del 50% en el riesgo de ventilación mecánica (OR= 0,50; IC95% 0,23-1,07) y del 44% en el riesgo de muerte (OR= 0,56; IC95% 0,19-1,66). Respecto al perfil toxicológico, se reportó una frecuencia de eventos adversos significativamente menor en el grupo de colchicina que en el grupo placebo (4,9% vs. 6,3%; p= 0,05), notificándose con menor frecuencia, por ejemplo, casos de neumonía (2,9% vs. 4,1% en el grupo control), si bien la diarrea fue más común en el grupo del fármaco (13,7% vs. 7,3%; p< 0,0001). Se debe recordar, no obstante, que la colchicina tiene un margen terapéutico estrecho y es extremadamente tóxica en caso de sobredosis, lo que requiere un ajuste muy riguroso de su pauta posológica. Ha consistido éste en un estudio aleatorizado, doble ciego y controlado por placebo (N= 1.033), que evaluó si baricitinib (4 mg/día, durante un máximo de 14 días) modificaba el tiempo que tardaban los pacientes con COVID-19 moderada-grave tratados con remdesivir –siempre junto con los cuidados estándar de soporte– en recuperarse clínicamente; se definió la recuperación como: alta hospitalaria o continuación de la hospitalización sin necesitar O2 suplementario ni atención médica continua. Tras un seguimiento durante 29 días, la mediana del tiempo hasta la recuperación para los pacientes que recibieron la combinación de fármacos (N= 515) se redujo en un 12,5%: fue de 7 días, frente a 8 días para los que recibieron placebo más remdesivir (N= 518); había un 30% más de probabilidades de mejoría clínica al día 15 en quienes recibieron baricitinib (OR= 1,3; IC95% 1,01-1,32; p= 0,03). El acortamiento del tiempo hasta la recuperación era mayor incluso en pacientes que recibían O2 de alto flujo o ventilación no invasiva al inicio (mediana de 10 días vs. 18 días en el grupo control; RR= 1,51; IC95% 1,10-2,08). También se observó una tendencia favorable para la mortalidad a 28 días: del 5,1% en el grupo de la combinación frente al 7,8% en el grupo control (HR= 0,65; IC95% 0,39-1,09). En relación con la seguridad, los posibles eventos secundarios asociados a la combinación (tasa de incidencia del 41% vs. 48% con remdesivir solo; 16% vs. 21% de eventos graves) incluyen los ya conocidos para los fármacos aislados: riesgo de infecciones graves, eventos trombóticos, alteración de pruebas de laboratorio y reacciones alérgicas. Aún se sigue evaluando el perfil beneficio-riesgo de la combinación, y cabe subrayar que baricitinib no ha sido aún autorizado en la UE para su uso en COVID-19.
Ha consistido éste en un estudio aleatorizado, doble ciego y controlado por placebo (N= 1.033), que evaluó si baricitinib (4 mg/día, durante un máximo de 14 días) modificaba el tiempo que tardaban los pacientes con COVID-19 moderada-grave tratados con remdesivir –siempre junto con los cuidados estándar de soporte– en recuperarse clínicamente; se definió la recuperación como: alta hospitalaria o continuación de la hospitalización sin necesitar O2 suplementario ni atención médica continua. Tras un seguimiento durante 29 días, la mediana del tiempo hasta la recuperación para los pacientes que recibieron la combinación de fármacos (N= 515) se redujo en un 12,5%: fue de 7 días, frente a 8 días para los que recibieron placebo más remdesivir (N= 518); había un 30% más de probabilidades de mejoría clínica al día 15 en quienes recibieron baricitinib (OR= 1,3; IC95% 1,01-1,32; p= 0,03). El acortamiento del tiempo hasta la recuperación era mayor incluso en pacientes que recibían O2 de alto flujo o ventilación no invasiva al inicio (mediana de 10 días vs. 18 días en el grupo control; RR= 1,51; IC95% 1,10-2,08). También se observó una tendencia favorable para la mortalidad a 28 días: del 5,1% en el grupo de la combinación frente al 7,8% en el grupo control (HR= 0,65; IC95% 0,39-1,09). En relación con la seguridad, los posibles eventos secundarios asociados a la combinación (tasa de incidencia del 41% vs. 48% con remdesivir solo; 16% vs. 21% de eventos graves) incluyen los ya conocidos para los fármacos aislados: riesgo de infecciones graves, eventos trombóticos, alteración de pruebas de laboratorio y reacciones alérgicas. Aún se sigue evaluando el perfil beneficio-riesgo de la combinación, y cabe subrayar que baricitinib no ha sido aún autorizado en la UE para su uso en COVID-19.

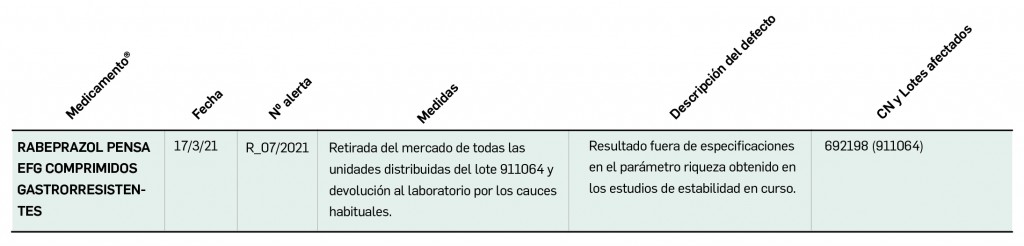
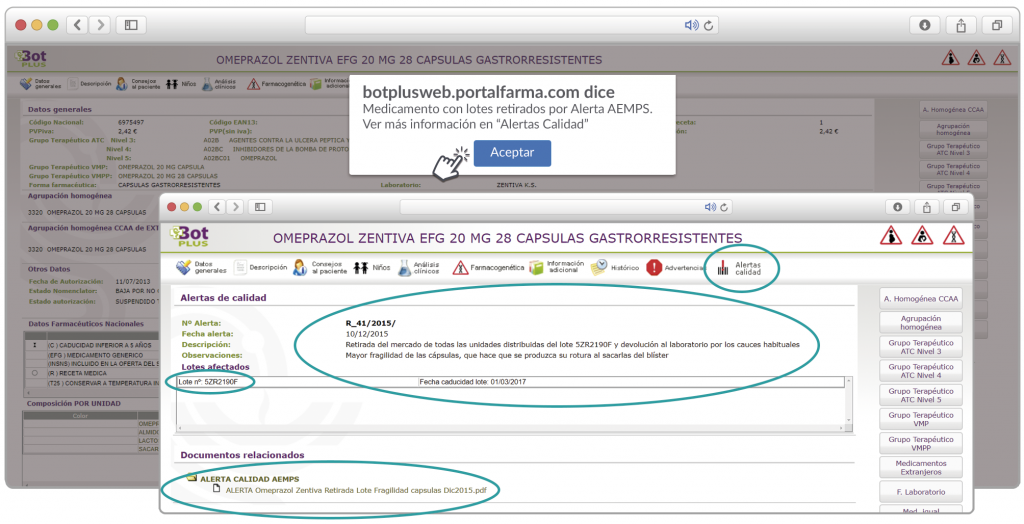
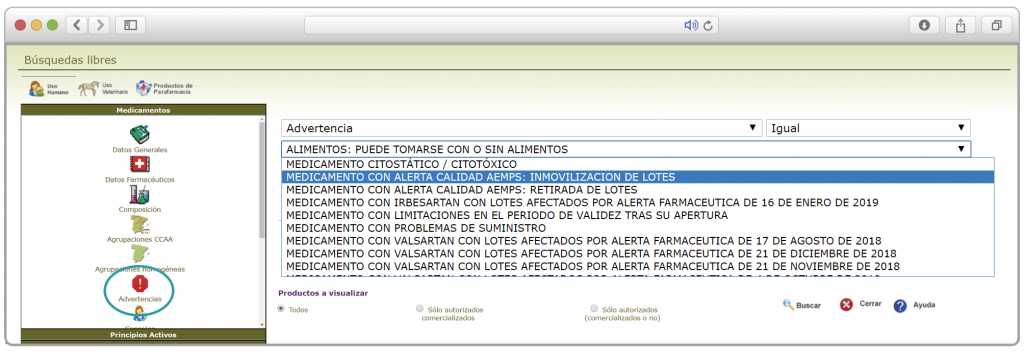
 Además, se incluye una pestaña específica en la que se pueden consultar los lotes concretos que han sido retirados, con sus respectivas fechas de caducidad, así como la descripción del defecto de calidad detectado y las medidas a adoptar. También se cuenta con acceso al documento publicado por la AEMPS.
Además, se incluye una pestaña específica en la que se pueden consultar los lotes concretos que han sido retirados, con sus respectivas fechas de caducidad, así como la descripción del defecto de calidad detectado y las medidas a adoptar. También se cuenta con acceso al documento publicado por la AEMPS.