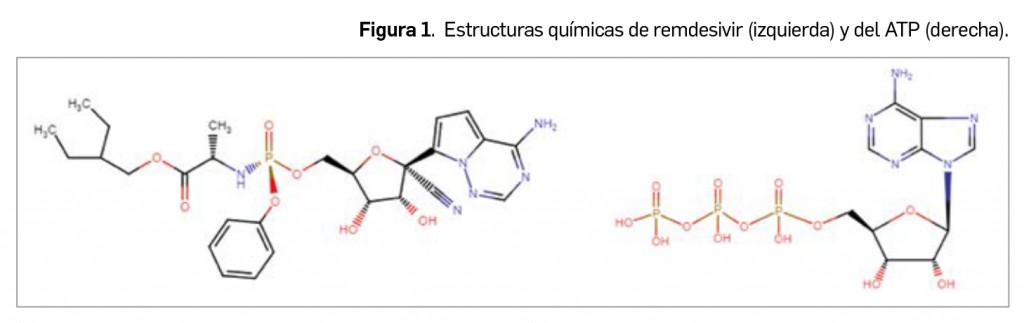
La falta de adherencia al tratamiento en pacientes que han sufrido un infarto de miocardio es un hecho común, no carente de eventos adversos. Las recomendaciones actuales abogan por la administración de inhibidores del receptor plaquetario P2Y12 (como clopidogrel o ticagrelor) durante, al menos, un año tras el infarto. Un cese prematuro de este tratamiento puede conducir eventos adversos cardiovasculares graves (EACG) como trombosis en el stent e, incluso, un nuevo infarto. Un factor asociado con la falta de adherencia y hasta el abandono del tratamiento es el coste del mismo, por lo que, en EE.UU., muchos hospitales facilitan el tratamiento para 30 días al alta del paciente; a pesar de ello, la falta de adherencia continúa siendo un problema.
El estudio ARTEMIS (Affordability and Real-World Antiplatelet Treatment Effectiveness After Myocardial Infarction Study) evaluó la adherencia al tratamiento y la incidencia de EACG en pacientes a los que se facilitó el tratamiento al alta durante 30 días respecto de una intervención en donde los pacientes no tuvieron copago durante un año tras el alta hospitalaria. Los resultados pusieron de manifiesto que, si bien la intervención aumentó la adherencia, no redujo la incidencia de eventos adversos. Se planteó entonces la hipótesis de si los hospitales que previamente disponían de programas de entrega gratuita del tratamiento obtendrían una mejor adherencia y persistencia tras el alta post-infarto que la de los hospitales que no disponían de tales programas. En aquel caso, sería menos probable que esos hospitales presentaran un beneficio superior que los hospitales sin previa entrega gratuita del tratamiento.
El estudio evaluó un total de 9.590 pacientes, 5.051 de 129 hospitales con programas gratuitos previos y 4.539 de 133 hospitales sin ellos. Asimismo, los pacientes de cada grupo fueron subdivididos aleatoriamente en un grupo control (con entrega de un mes de tratamiento) o en el de intervención (sin copago durante un año). Las variables principales fueron la persistencia al inhibidor del P2Y12 y la incidencia de EACG.
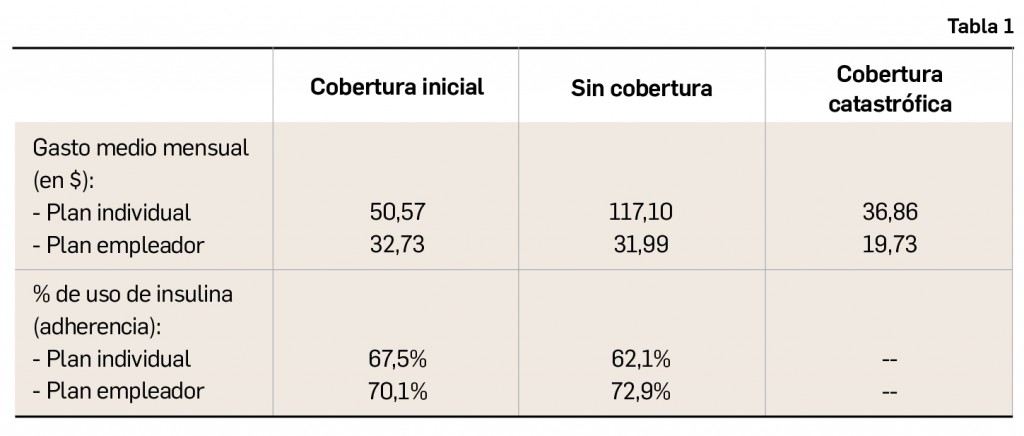
Los datos (Tabla 2) revelan que la persistencia media al tratamiento fue del 96% a los 90 días y del 52% al año. Los hospitales con programas gratuitos previos al estudio no mostraron diferencia en la persistencia a corto y largo plazo o con la incidencia de eventos. Sin embargo, la intervención sobre el copago incrementó la persistencia a un año en los dos tipos de hospitales –con y sin planes previos– pero no mostró una diferencia significativa en la incidencia de EACG en los dos grupos de hospitales.
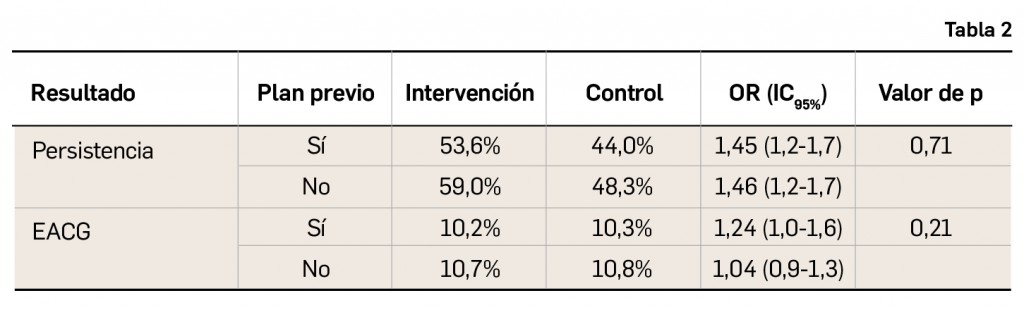
A la vista de los resultados, los autores concluyen que no se observó ninguna asociación entre la provisión gratuita del tratamiento con inhibidores de P2Y12 con la persistencia o la incidencia de eventos adversos cardiovasculares graves en aquellos pacientes que disponen de un seguro de cobertura de tratamientos. Por tanto, una intervención que anulaba el copago durante un año resultó significativamente exitosa respecto de la mejora de la persistencia al tratamiento, tanto en hospitales que disponían de planes para reducir los costes de los fármacos, como en aquellos que no disponían de los mismos.
Este estudio pone de manifiesto, una vez más, el enorme problema que supone la falta de adherencia y persistencia a los tratamientos. El coste de los mismos constituye una gran barrera, especialmente en las capas sociales más deprimidas económicamente. La actual situación de pandemia, con enormes repercusiones en la economía, tanto macro como micro, permite intuir que este problema sucede también en nuestro país, por lo que los farmacéuticos deberían estar vigilantes al respecto e implementar acciones que traten de contrarrestar la discontinuación de los tratamientos por cualquier causa.