La autorización condicional en Europa de las diversas vacunas frente a la infección por coronavirus SARS-CoV-2 establece un seguimiento de todas y cada una de las nuevas informaciones sobre su seguridad. Las agencias nacionales reguladoras de la Unión Europea, como la AEMPS, informan periódicamente de manera coordinada junto con la EMA (Agencia Europea de Medicamentos). Igualmente, la Organización Mundial de la Salud (OMS), como otras organizaciones, aportan su evaluación de la seguridad de las vacunas COVID-19.
Las 27 agencias nacionales europeas, junto con la Agencia Europea de Medicamentos (EMA), mantienen una evaluación continua de todos los eventos adversos relacionados con la vacunación frente a la COVID-19, sean atribuidos a la vacuna o al proceso de vacunación, por posibles errores programáticos. En la Unión Europea, el sistema integral de control de la seguridad y gestión de riesgos (farmacovigilancia) de las vacunas frente a la COVID-19 se basa en las evaluaciones mensuales del comité europeo Pharmacovigilance Risk Assessment Committee (PRAC), con 35 expertos, y del COVID-19 EMA pandemic Task Force (COVID-ETF) que garantizan:
- brindar asesoramiento para minimizar el riesgo;
- notificar sospechas de reacciones adversas a los medicamentos (RAM) de los profesionales y ciudadanos;
- realizar estudios después de la autorización (EPAS);
- detectar posibles RAM en colaboración con los programas de
- vacunación;
- realizar evaluaciones científicas rigurosas de todos los datos de seguridad;
- introducir las acciones de mitigación necesarias.
Una de las actividades concretas es la publicación cada mes de las evaluaciones de los problemas de seguridad o safety updates que el PRAC lleva a cabo con los informes periódicos de seguridad (IPS) mensuales que los laboratorios titulares de cada vacuna deben presentar en la EMA, como requisito de la “autorización condicional”.
Así, el 25 de febrero de 2021, después de evaluar nuevos datos de seguridad, el PRAC concluyó que el balance beneficio-riesgo de las vacunas Comirnaty® (Pfizer/BioNTech) y COVID-19 Vaccine Moderna® (Moderna) permanecen sin cambios. La evaluación cubrió todos los nuevos datos de seguridad que surgieron en todo el mundo, incluido el segundo Informe de seguridad mensual resumido y una revisión de las reacciones alérgicas graves del titular de la autorización de comercialización (TAC). Los TAC de las vacunas COVID-19 compilarán informes de seguridad mensuales resumidos a fin de respaldar las evaluaciones de riesgo-beneficio oportunas y continuas. La presentación de dichos informes complementa la presentación de los IPS actualizados.
Específicamente, el PRAC concluyó lo siguiente en relación con las siguientes RAM:
Diarrea y vómitos
La evaluación identificó la diarrea y los vómitos después de la vacunación como nuevos efectos secundarios de Comirnaty®. La frecuencia de estos efectos secundarios y el grado en que ocurren se están evaluando más a fondo. La información del producto se actualizará en consecuencia.
Reacción alérgica grave (Anafilaxia)
Se evaluaron los informes de casos de sospecha de anafilaxia para Comirnaty® y COVID-19 Vaccine Moderna®, tras lo cual no se produjeron cambios en el uso recomendado de estas vacunas. La anafilaxia continúa siendo monitorizada de cerca. La información sobre el tratamiento clínico de la anafilaxia ya está disponible en la información oficial de las dos vacunas.
Sospecha de efectos secundarios con desenlace fatal
El PRAC evaluó informes de casos de sospechas de efectos secundarios con desenlace fatal después de la vacunación con Comirnaty® y COVID-19 Vaccine Moderna®. En la mayoría de los casos, la progresión de (múltiples) enfermedades preexistentes parecía ser una explicación plausible de la muerte. En algunas personas, los cuidados paliativos se iniciaron antes de la vacunación. Esta evaluación de los datos disponibles no lo identificó como un problema de seguridad.
Casos mortales en Noruega
Como complemento a lo descrito anteriormente, una revisión periódica de la Organización Mundial de la Salud (OMS) de los casos notificados a nivel mundial, publicada el 14 de enero de 2021, evaluó los 23 casos mortales notificados en Noruega en conexión con la vacunación en adultos mayores severamente frágiles. Para esa fecha, 43.740 personas habían sido vacunadas contra la COVID-19 en aquél país, con un número importante de vacunados residentes en residencias de mayores. El Subcomité del Global Advisory Committee on Vaccine Safety (GACVS) de la OMS, en su reunión del 19 enero, revisó la información disponible y concluyó que no sugiere ningún aumento inesperado o desfavorable de las muertes en personas frágiles, ancianos o cualquier característica inusual de eventos adversos después de la administración de la vacuna Comirnaty® (Pfizer/BioNTech). Ya el 2 de febrero, ascendió la cifra a 53 muertes en la población de adultos mayores, en un total de 112.080 vacunados en el país. Es preciso recordar que los adultos mayores en residencias tienen un riesgo más elevado de mortalidad por sus condiciones severas de base, siendo la expectativa de vida más corta que en la población general. Se contempla, en promedio, que cada semana mueren en Noruega alrededor de 300 personas de residencias de personas mayores.
Casos de parálisis facial en Reino Unido
La agencia reguladora británica, Medicines and Healthcare Products Regulatory Agency (MHRA), ha recibido hasta el 31 enero 2021 un total de 99 notificaciones de sospechas de parálisis facial, Bell’s palsy o paresia (parálisis de Bell), asociadas a la vacuna Comirnaty® (Pfizer/BioNTech). Este evento se indica actualmente como un posible evento adverso relacionado con la inmunización con la vacuna Comirnaty®, en base al pequeño número de notificaciones reportadas en los ensayos clínicos, pero como este efecto también puede ocurrir naturalmente, aún no está confirmada su asociación con la vacuna.
Dicha agencia también ha recibido 15 notificaciones de sospechas de parálisis facial para la vacuna COVID-19 Vaccine AstraZeneca®.
Además de la revisión clínica individual, se están analizando estas notificaciones en función del número de casos que se esperaría que ocurrieran en ausencia de vacunación, esto es, la “incidencia basal” o “tasa natural” de ocurrencia por cualquier causa. Sobre la base de esta revisión, el número de notificaciones de parálisis facial recibidos hasta ahora es similar a la tasa natural esperada, y actualmente no sugiere un mayor riesgo con las vacunas. No obstante, se continuarán monitorizando estos “eventos adversos de especial interés” (AESI, por sus siglas en inglés), incluso a través de la evaluación de los datos electrónicos del registro de atención médica, dada su trascendencia en el perfil de seguridad, objetivo de la fase de seguimiento en situaciones habituales de la farmacovigilancia.
Casos de trombocitopenia inmune en EE. UU.
De acuerdo con The New York Times, ha habido 36 notificaciones de personas que recibieron las vacunas Comirnaty® o COVID-19 Vaccine Moderna® y que poco después desarrollaron el poco frecuente trastorno sanguíneo “trombocitopenia inmune”, con cuadros hemorrágicos. Un equipo de expertos de la Food and Drug Administration (FDA) y del Centro para el Control y la Prevención de Enfermedades (CDC) de EE.UU. están investigando estos casos y han señalado que los casos podrían ser coincidentes en el tiempo y no estarían asociados con las vacunas utilizadas.
En los estudios clínicos realizados en fase 3 con estas vacunas, y que involucraron aproximadamente unas 30.000 y 44.000 personas (de Moderna y de Pfizer/BioNTech, respectivamente), no se reportó ningún evento adverso de “trombocitopenia inmune”. Según la FDA, cada año en los EE.UU. se notifica alrededor de 1 caso con esta condición por cada 35.000 personas.
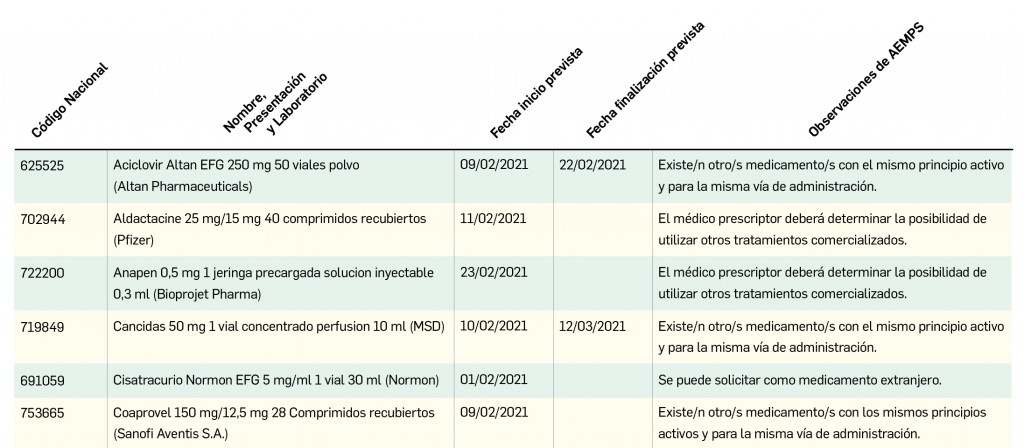
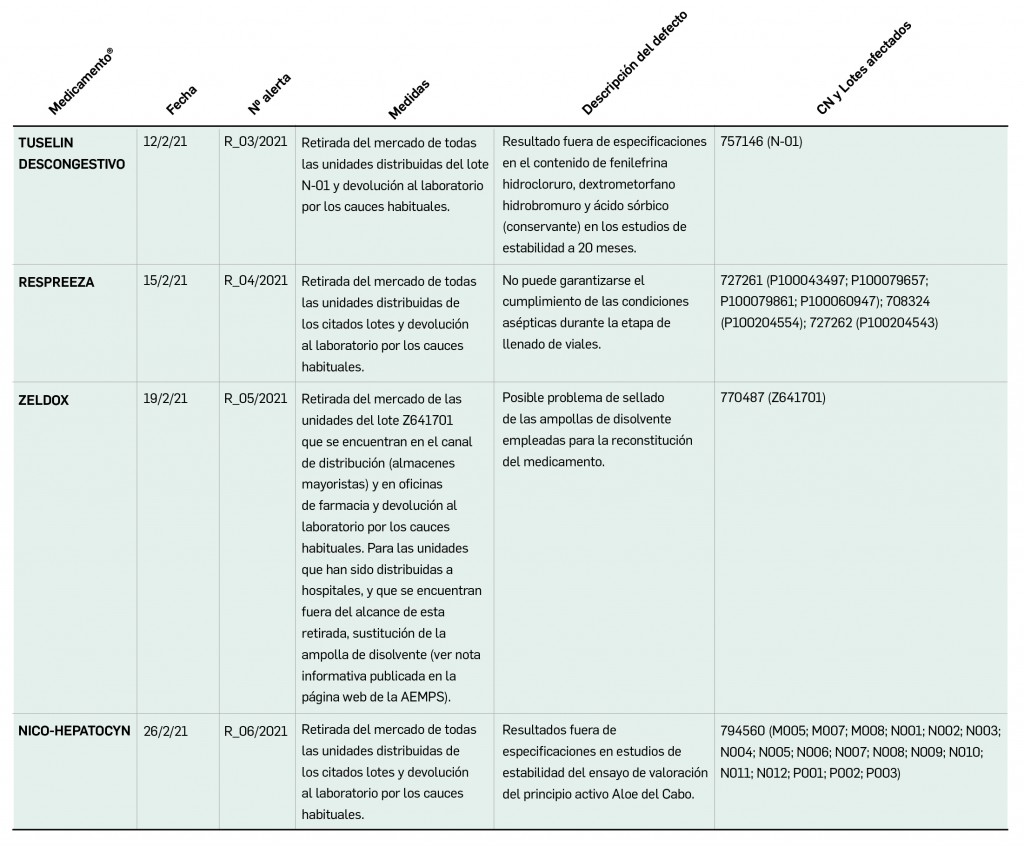
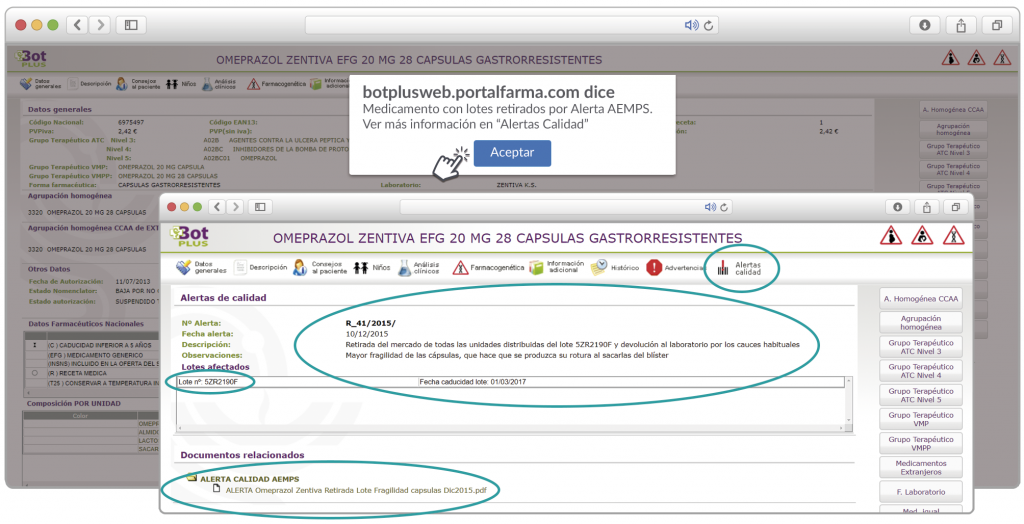
 Además, se incluye una pestaña específica en la que se pueden consultar los lotes concretos que han sido retirados, con sus respectivas fechas de caducidad, así como la descripción del defecto de calidad detectado y las medidas a adoptar. También se cuenta con acceso al documento publicado por la AEMPS.
Además, se incluye una pestaña específica en la que se pueden consultar los lotes concretos que han sido retirados, con sus respectivas fechas de caducidad, así como la descripción del defecto de calidad detectado y las medidas a adoptar. También se cuenta con acceso al documento publicado por la AEMPS.