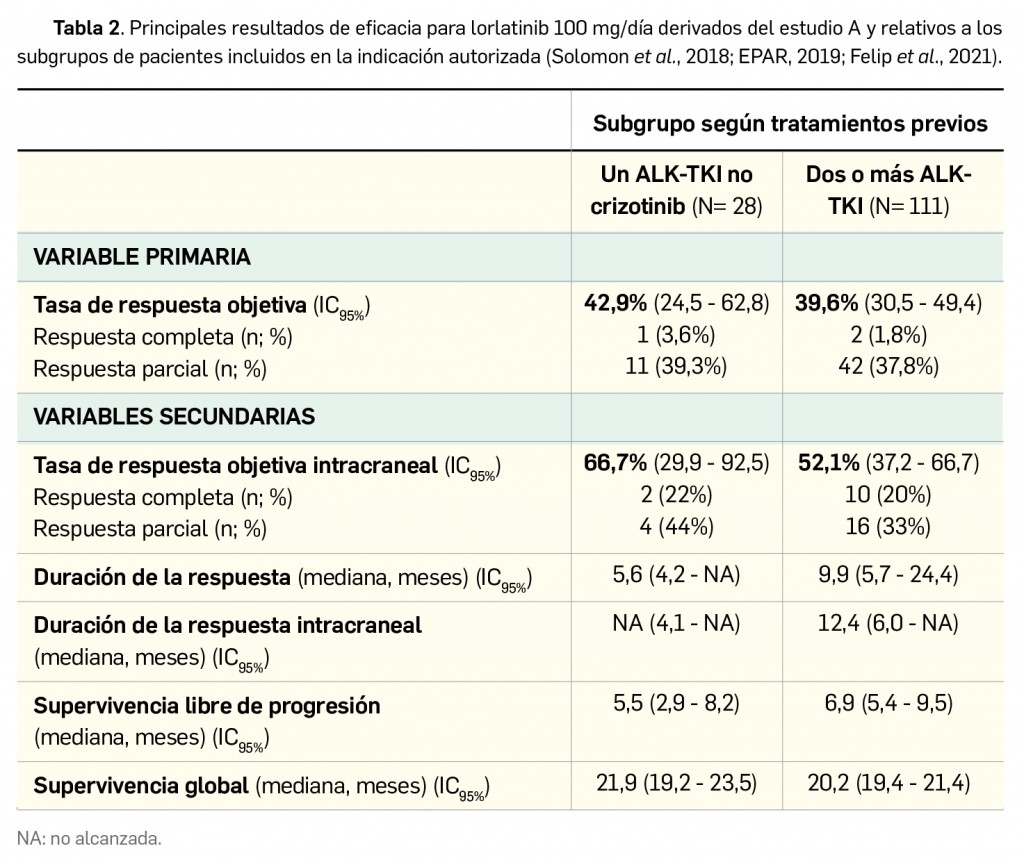
Resumen
El cólera, enfermedad infecciosa causada por ciertos serotipos del bacilo Vibrio cholerae, representó una epidemia de importantes magnitudes en diversas zonas de España a finales del siglo XIX. En concreto, una Real Orden que declaró oficialmente su presencia –de unos pocos casos– en la ciudad de Madrid fue publicada el 16 de junio de 1885. Las medidas sanitarias tomadas por las autoridades locales, entre ellas la depuración de las aguas, fumigaciones o aislamiento de los enfermos, desencadenaron una serie de protestas por parte de la población madrileña como consecuencia, entre otras, del impacto negativo que tales medidas podrían tener sobre el comercio local.
El presente artículo aborda oportunamente –dada la situación actual derivada de la pandemia de COVID-19– una revisión histórica del contexto socio-político y sanitario en esos meses turbulentos del Madrid de 1885, y reproduce algunas de las principales protestas y motines que por entonces acontecieron contra la declaración del cólera en la capital. Habida cuenta de que la mayoría de los episodios de rebelión pública en torno al cólera los protagonizaron mujeres, se ilustra con curiosas imágenes que ponen de manifiesto el destacado papel que la figura de la mujer ha tenido en la publicidad farmacéutica, sobre todo, a lo largo del siglo XX.
Nota del autor
Hace algunos años ya, Benito del Castillo y yo publicamos un libro sobre la mujer en la publicidad farmacéutica1, con carteles y postales de ambas colecciones. Desde entonces, la nuestra ha crecido mucho. Voy a comenzar a ocuparme de las mujeres en la publicidad farmacéutica mediante un artículo sobre el protagonismo de las féminas en algunas de las grandes e injustas protestas organizadas en Madrid ante la declaración del cólera en 1885. Anécdotas precursoras de las negacionistas, padecidas ante la COVID, explicativas de cómo –cuando la naturaleza nos ataca– el miedo, o incluso el pánico, pone en funcionamiento lo más irracional de nosotros mismos y del gran impedimento para el comercio y la economía que siempre han supuesto las epidemias y, en otro aspecto, el protagonismo femenino entre el pueblo llano por muy variadas razones (Figura 1).
En las ilustraciones de este artículo emplearé propaganda, principalmente de farmacias, de diversos lugares de España.
 La declaración oficial de la existencia del cólera
La declaración oficial de la existencia del cólera
Su presencia en Valencia, Castellón, Murcia y la capital se declaró oficialmente por una Real Orden, publicada en la Gaceta de 16 de junio de 1885. Con respecto a Madrid, se indicaba la existencia de pocos casos, la esperanza de poderlos controlar y se justificaba la publicación de la disposición legal para contribuir a mantener la confianza de un lado y hacer imposible de otro falsos rumores que difundan injustificadas alarmas2. Aunque algunos historiadores retrotraen su aparición al día 4 del mismo mes3, en realidad la epidemia se inició, como tarde, el 20 de mayo4.
Pese a lo retardado de la declaración oficial, la oposición a la misma fue frontal. En El Correo Militar, acaso el periódico menos crítico, se daba cuenta de un nuevo padecimiento: la enfermedad sospechosa, pero se habían librado del cólico, producido por comer fruta verde y barata: si lo que hay en Madrid es cólera –escribía– no es cólera morbo asiático, sino el cólera de los pobres.
En La Higiene, del mismo día, se aseguraba haber disminuido la alarma producida por los casos sospechosos, lo cual era excelente para ahuyentarel miedo, tan perturbador de todas las funciones, especialmente las digestivas5. En el periódico liberal El Día, se asombran de la declaración, pues Romero Robledo, ministro de la Gobernación del Gobierno conservador de Cánovas del Castillo, había dicho ocho días antes en el Congreso que acaso se tratara del llamado cólico de Madrid. A su parecer, con la misma, no llevaban la tranquilidad a la población dada la disparidad de criterios existentes entre los médicos. Todavía el 26 de junio consideraban improcedente la declaración, hubiera o no cólera en Madrid6.
En La Iberia. Diario Liberal se escribió: por fin se ha dado en Madrid el golpe de muerte que venía amenazándola desde hace algunos días; al fin se ha consumado la obra de matar nuestro comercio, porque ayer hubo siete cólicos en una población de 500.000 almas; al fin el Gobierno ha hecho la última calaverada. Al tiempo que excitaba, directamente, a la protesta de los industriales y comerciantes7.
En El Día del 17 de junio, se incluyó en la portada un artículo titulado La cólera contra el comercio, en donde se acusa al Gobierno de llevar al país a la completa ruina a costa de las teóricas medidas sanitarias, pues los gobernadores y autoridades municipales, por la circular del 12 de junio, quedaron autorizadas para instalar acordonamientos y lazaretos para las provincias limítrofes; al autorizar uno en Madrid, para gentes procedentes de Valencia, se establecía un precedente muy grave a ojos de los comerciantes8.
En El Siglo Futuro, diario fundado por Cándido Nocedal y ligado al integrismo y al carlismo, motejaron a la declaración oficial de terrorismo epidémico9. En El Liberal, incluyeron un artículo en primera página titulado La mentira de la “Gaceta de Madrid”, en donde consideraban imprevisible la contestación de un pueblo por una declaración gubernamental que alarma, daña los intereses de los ciudadanos y suscita un debate apasionado en el parlamento, tan solo por la aparición de unos cuantos casos, no se sabe bien si de enfermedad manifiesta o simplemente sospechosos10 (Figura 2).
 La República se mostró igual de contundente pues, a su parecer, la declaración se efectuó sin verdadero motivo, sin fundamento serio, postura negacionista que, al revés del resto de la prensa, mantuvieron durante casi toda la epidemia, como también sostuvieron, mientras continuó en el Ministerio, una posición absolutamente beligerante con Romero Robledo, más dura que el resto de las publicaciones opositoras11.
La República se mostró igual de contundente pues, a su parecer, la declaración se efectuó sin verdadero motivo, sin fundamento serio, postura negacionista que, al revés del resto de la prensa, mantuvieron durante casi toda la epidemia, como también sostuvieron, mientras continuó en el Ministerio, una posición absolutamente beligerante con Romero Robledo, más dura que el resto de las publicaciones opositoras11.
En La Época, diario gubernamental, se asombran de las quejas producidas por la declaración oficial, a causa de los posibles perjuicios causados al comercio y al mercado internacional, cuando ya había sido declarada la “procedencia sucia” –que impedía las exportaciones– por Francia, Italia, Portugal y Alemania, para las mercaderías procedentes de España, efectuada mucho antes de la declaración oficial. Se sorprenden de la teórica inconveniencia de proclamar en alta voz lo susurrado por lo bajo.
Así sucedió a lo largo de todo el periodo epidémico, pues los opositores no llegaron a aceptar la existencia de la enfermedad, pese al número afortunadamente no muy elevado de fallecidos.
Las primeras protestas: Santa Casilda, San Idelfonso, las lavanderas, las cigarreras y los pobres
El Ayuntamiento madrileño había empezado con las medidas anticoléricas, sottovoce, mucho antes de la declaración oficial del cólera en la capital. A partir del 16 de junio las disposiciones fueron públicas y mucho más enérgicas.
Conviene recordar que la cifra de fallecidos en Madrid, según la Memoria de Alberto Bosch, fue de 1.366 personas, el 0,27% de la población (por los comentarios efectuados en los periódicos a los que hemos hecho referencia la población se acercaba a los 500.000 habitantes), el menor para una ciudad en constante crecimiento, y su paso fue menos devastador que en otros lugares del país12. Se han dado diversas explicaciones para el fenómeno, a mi parecer se debió a la enérgica actuación del Alcalde, magníficamente asesorado por Fausto Garagarza. El director del laboratorio municipal no era médico. Sí un magnífico farmacéutico, buen microscopista y especializado en el análisis de las aguas. Se desentendió de las disputas médico-teóricas sobre el origen colérico de la enfermedad, pero conocía bien lo mantenido por Koch. Por ello sabía, o creía saber, que el bacilo Vibrio cholerae era el causante de la enfermedad; se transmitía por las aguas y estaba en las deyecciones y aguas negras de los contagiados. No sé si se lo expuso de esa manera o no al Alcalde. Fuera como fuese, enfocaron su labor a abastecer Madrid con aguas sin el bacilo y publicaron diariamente el estado microbiológico y químico de las mismas.
 La segunda medida fue la utilización de las fumigaciones, con la intención de matar al agente etiológico. Lo hicieron a mansalva con algunos productos que desprendían cloro, el elemento utilizado en la actualidad para sanear las aguas de consumo. Si bien su empleo sobre las personas es probable que no fuera demasiado eficaz, el realizado sobre las aguas negras sí lo sería, aunque tal vez no lo suficientemente exhaustivo y profundo como para acabar con todos los focos. La tercera medida tomada fue el aislamiento de los enfermos de una manera rígida y vigilada por agentes del orden (Figura 3).
La segunda medida fue la utilización de las fumigaciones, con la intención de matar al agente etiológico. Lo hicieron a mansalva con algunos productos que desprendían cloro, el elemento utilizado en la actualidad para sanear las aguas de consumo. Si bien su empleo sobre las personas es probable que no fuera demasiado eficaz, el realizado sobre las aguas negras sí lo sería, aunque tal vez no lo suficientemente exhaustivo y profundo como para acabar con todos los focos. La tercera medida tomada fue el aislamiento de los enfermos de una manera rígida y vigilada por agentes del orden (Figura 3).
Las medidas no fueron entendidas, en su momento, por casi nadie. La discusión sobre los microbios es ridícula vista desde una mentalidad del primer tercio del siglo XXI, pero no en su momento, y muchos de los habitantes pobres de Madrid eran analfabetos. Sí sabían todos cómo la epidemia se encarnizaba con los pobres. Conocían la falta de eficacia de las medidas anteriores. Estaban al tanto de la ausencia de medicamentos capaces de sanarles. Desconfiaban de la farmacología al uso, pues además de ineficaz era atormentadora. Las clases desfavorecidas oían hablar, desde el primer brote, de la necesidad de hacer más higiénica la ciudad, sin resultado alguno. Veían como se aplicaba la caridad sobre ellos mientras duraba la epidemia y luego volvían al olvido. Tenían la experiencia de que las medidas anti-epidémicas las sufrían ellos por encima de cualquier otra persona o grupo social, por eso debe relativizarse cuando se lee sobre la incultura de las gentes en la prensa de la época. Eran incultos, sí, e incluso analfabetos muchos de quienes se levantaron contra las medidas anti epidémicas, pero en su incultura y analfabetismo tenían una larga experiencia de sufrimiento, durante los contagios, mucho mayor en ellos que en el resto de las clases sociales madrileñas.
EL MOTÍN DEL PARADOR DE SANTA CASILDA: ¡NO QUIERO POLVOS, SINO PAN PARA MIS HIJOS!
El así llamado era un tejar situado cerca de la Puerta de Toledo. Tenía dos pisos y multitud de pasillos conducentes a las habitaciones interiores; en ellas se alojaban más de trescientos vecinos. Benito Pérez Galdós lo describe de esta manera: no lejos del punto en que Mesón de Paredes desemboca en la Ronda de Toledo, hallaron el parador de Santa Casilda, vasta colmena de viviendas baratas alineadas en corredores sobrepuestos. Entrase a ella por un patio o corralón largo y estrecho, lleno de montones de basura, residuos, despojos y desperdicios de todo lo humano…13
El laboratorio municipal envió allí varios carros fumigadores, cargados de ácido fénico y cloruro cálcico, el día 16 de junio. En cuanto los habitantes vieron de lejos los carros de los microbios, como los llamaban las gentes madrileñas, unas cien mujeres se colocaron en la puerta para impedir su acceso, algunas con los chiquillos colgándoles del pecho, mientras gritaban: ¡Aquí no queremos microbios! ¡Que se vayan, que se vayan!
 Los empleados municipales enviaron aviso al Alcalde de lo sucedido. Alberto Bosch se presentó en el parador y trató de explicar a las mujeres cual era el objeto de la fumigación. No consiguió nada. Solo más gritos, ahora algo mejor dirigidos: ¡no queremos veneno! ¡Que se vayan! Frustrado en su misión, el Alcalde avisó al Gobernador, quien mandó al coronel Oliver con varios guardias. Todos ellos trataron, de nuevo, de persuadir a las mujeres. Su misión fue contraproducente pues los ánimos se encocoraron contra la fuerza pública. Una de las mujeres se acercó al coronel Oliver y le dijo: No quiero polvos, sino pan para mis hijos (Figura 4).
Los empleados municipales enviaron aviso al Alcalde de lo sucedido. Alberto Bosch se presentó en el parador y trató de explicar a las mujeres cual era el objeto de la fumigación. No consiguió nada. Solo más gritos, ahora algo mejor dirigidos: ¡no queremos veneno! ¡Que se vayan! Frustrado en su misión, el Alcalde avisó al Gobernador, quien mandó al coronel Oliver con varios guardias. Todos ellos trataron, de nuevo, de persuadir a las mujeres. Su misión fue contraproducente pues los ánimos se encocoraron contra la fuerza pública. Una de las mujeres se acercó al coronel Oliver y le dijo: No quiero polvos, sino pan para mis hijos (Figura 4).
Tras seis horas sin llegar a acuerdo alguno, los carros del laboratorio municipal se retiraron si llevar a cabo su labor14. El día 18 los vecinos permitieron la fumigación sin problema de ningún tipo. El Gobernador se enteró de la necesidad de la mayoría, de sus dificultades para comprar comida, y les dio dinero. Todos los insultos de los días anteriores se tornaron en alabanzas15.
Este hospedaje estaba situado en las afueras del Madrid de los barrios bajos, de la capital oscura y suburbial. Un periodista de El Liberal, Julio Vargas Machuca, escribió una serie de artículos, titulados Madrid ante el cólera, en donde se da cuenta del reverso de lo efectuado por Ramón Mesonero Romanos en su obra. En un espacio periodístico breve, se da fe de la forma de vida, o mejor de subsistencia, de las clases madrileñas más desfavorecidas, en un ambiente higiénico-sanitario apocalíptico, más dramáticamente expresado que en los textos de los higienistas16.
Para entender la vida en los barrios bajos, podemos leer la novela de Pérez Galdós antes mencionada o a Pío Baroja. El último, en La busca, nos deja un testimonio prístino sobre la existencia de “los madriles”: El madrileño que alguna vez, por casualidad, se encuentra en los barrios pobres próximos al Manzanares, hállase sorprendido ante el espectáculo de miseria y sordidez, de tristeza e incultura que ofrecen las afueras de Madrid con sus rondas miserables, llenas de polvo en verano y de lodo en invierno. La corte es ciudad de contrastes; presenta luz fuerte al lado de sombra oscura; vida refinada, casi europea, en el centro; vida africana, de aduar, en los suburbios17.
Ante la situación descrita, no puede sorprendernos lo sucedido en el parador de Santa Casilda, tampoco el amotinamiento del vecindario durante el mes de agosto; para sofocarlo fue necesaria la presencia de las fuerzas de orden público18.
LAS VERDULERAS DEL MERCADO DE SAN ILDEFONSO
Edificado en 1835, ocupaba la plazuela del mismo nombre en su totalidad. Construido en hierro, tenía una preciosa estampa, similar a la del desaparecido de la Plaza de Olavide o al todavía conservado de San Miguel. Pese a su apertura, no se acabó la venta de productos alimenticios en la Corredera Alta y Baja, en el entorno del local cubierto. Cuando se desplegaban los puestos, se abrían como los pétalos de una flor y muchos de ellos daban a las calles circundantes. Una vez cerrados, se podía clausurar todo el recinto mediante puertas de rejas deslizantes. Así quedaba por las noches, bajo la custodia de los cientos de gatos habitantes de su tejado, al menos en la primera mitad del siglo XX, hasta la fecha de su derribo en 1970 (Figura 5).
 Tras los sucesos mencionados, el 17 de junio, los comerciantes vieron llegar hasta su lugar de trabajo los carros desinfectantes. Inmediatamente se parapetaron todos tras las verduleras, al grito de: ¡El cólera! ¡Nos traen el cólera! Los encargados de las mangueras se detuvieron ante la actitud de aquellas mujeres y sus gritos, según los cuales no queremos que nos maten con polvos como a las chinches, ¡que se los echen al Gobernador!
Tras los sucesos mencionados, el 17 de junio, los comerciantes vieron llegar hasta su lugar de trabajo los carros desinfectantes. Inmediatamente se parapetaron todos tras las verduleras, al grito de: ¡El cólera! ¡Nos traen el cólera! Los encargados de las mangueras se detuvieron ante la actitud de aquellas mujeres y sus gritos, según los cuales no queremos que nos maten con polvos como a las chinches, ¡que se los echen al Gobernador!
A causa del alboroto llegaron los guardias municipales. Todos los vendedores se encerraron en el mercado, sin deponer su actitud. Llegó el inspector del distrito, a quien apodaban El Chato. Pese a la familiaridad que presupone el mote, fue recibido con una silba espectacular. Conferenció con el administrador del establecimiento quien le dio fe de haberse tomado allí todas las precauciones higiénicas necesarias, por lo cual no estaba dispuesto a deponer su actitud; cuando se disponían a actuar por la fuerza, llegó una orden del Gobierno civil. Al recibirla, los carros fumigadores se batieron en retirada de nuevo19.
El mercado fue fumigado el día 18 sin mayores inconvenientes20.
LAS CIGARRERAS
Así se llamaba a las mujeres que trabajaban en la fábrica de tabacos. En los primeros días del mes de marzo de 1885, organizaron un gran motín ante la sospecha de la introducción de máquinas en la misma mediante las cuales disminuiría su carga de trabajo. Tuvo que intervenir la Guardia Civil y se organizó un enorme lío con heridos y detenidas, reproducido algunos días después en la fábrica de tabacos de Sevilla21. Este hecho las ha dado una gran fama de belicosas, no se ha contemplado en la mayoría de la bibliografía como un suceso ludista más, sino desde un prisma feminista y de conciencia de clase.
Aunque puede haber algo de eso, la mayoría de los episodios de rebelión pública en torno al cólera, como estamos viendo, los protagonizaron mujeres. Evidentemente tenían conciencia de sus derechos, pero también una gran carga de ignorancia impuesta por las limitaciones de su formación. Por otra parte, eran enviadas por delante en todas las reivindicaciones pues, aparte de vivir en una sociedad machista, el papel de las mujeres en las familias era muy importante. El desparpajo de las mujeres de clase trabajadora no tenía nada que ver con el de los hombres y, además, por ser una sociedad patriarcal, la represión ejercida por los cuerpos de seguridad sobre las mujeres era mucho más considerada y débil que al actuar sobre los hombres.
La cuestión de las cigarreras en la epidemia de cólera es mucho menos conocida porque no las deja en tan buen lugar como en el caso de su rebelión contra las máquinas.
 Entre las medidas anticoléricas propuestas por el Alcalde de Madrid, se planteó el establecimiento de un hospital provisional para los atacados, situado en la Escuela de Veterinaria de la capital. La misma estaba entonces situada en Embajadores, cerca de la fábrica de tabaco. Muchos de los habitantes del barrio elevaron una protesta al Ayuntamiento y al Gobierno civil; las cigarreras, con su fama belicosa empezaron con reproches públicos y ruidosos el mismo 16 de junio, al salir del trabajo22. Al día siguiente, cuando los operarios municipales se presentaron para fumigar la fábrica, las operarias rompieron los aparatos de fumigación y pusieron en fuga a los empleados. A su parecer, estaban suficientemente fumigadas con el tabaco23 (Figura 6).
Entre las medidas anticoléricas propuestas por el Alcalde de Madrid, se planteó el establecimiento de un hospital provisional para los atacados, situado en la Escuela de Veterinaria de la capital. La misma estaba entonces situada en Embajadores, cerca de la fábrica de tabaco. Muchos de los habitantes del barrio elevaron una protesta al Ayuntamiento y al Gobierno civil; las cigarreras, con su fama belicosa empezaron con reproches públicos y ruidosos el mismo 16 de junio, al salir del trabajo22. Al día siguiente, cuando los operarios municipales se presentaron para fumigar la fábrica, las operarias rompieron los aparatos de fumigación y pusieron en fuga a los empleados. A su parecer, estaban suficientemente fumigadas con el tabaco23 (Figura 6).
En la mañana del día veinte, un grupo de unas cien cigarreras se negaron a entrar al trabajo e intentaron manifestarse, pacíficamente, en la calle Embajadores. Alguna de ellas empezó a insultar a los guardias y las dispersaron. Se volvieron a concentrar en el otro extremo de la calle y volvieron a disolver su grupo. Pese a los sucesivos incidentes no se desanimaron. Otra vez se reunieron en el Puente de Toledo y a las doce del mediodía dejaron su intento de manifestarse por todo Madrid24. Nada dicen los periodistas de sus motivos pero, dados los antecedentes de protesta contra las fumigaciones y el establecimiento de un hospital cercano para los enfermos de cólera, añadidos al malestar general por la declaración del cólera y la manifestación de protesta convocada por los comerciantes, como si el Gobierno fuera el causante de la enfermedad, sus motivaciones parecen evidentes, si bien El Imparcial, seguramente consciente de lo impropio de las causas, vuelve a hacerse eco de las anteriores reivindicaciones contra las máquinas de fabricar cigarrillos25.
LAS LAVANDERAS
El día 18 de junio ocurrió un caso tragicómico. Afortunadamente no acabó en motín, pero estuvo a punto.
Al final de la calle Segovia, el día 11 del citado mes hubo una serie de casos considerados sospechosos de cólera y una defunción. Las medidas tomadas fueron fumigar el domicilio familiar, quemar las ropas de la fallecida y dejar aislada a toda la familia. Así las cosas, sin presentarse ninguna otra invasión colérica, uno de los aislados burló la vigilancia de los guardias de orden público. Corriendo a todo correr se dirigió hacia el puente de Segovia, seguido por los guardias, sable en mano. El vecindario se alborotó y tomó partido por el huido. Todas las lavanderas subieron desde el Manzanares hasta el puente de Segovia y no cesaron de zaherir a los guardias –en frase del periodista– con palabras pintorescas y picantes.
Ante la situación, telegrafiaron al Gobernador quien dispuso la puesta en libertad del detenido –a quien habían apaleado concienzudamente con anterioridad ante el disgusto de las lavanderas– y el cese del aislamiento del resto de la familia26.
LAS PROTESTAS DE LOS COMERCIANTES
El mismo martes, 16 de junio, nada más conocerse la declaración oficial de cólera, se reunió la junta directiva del Círculo de la Unión Mercantil, en principio para estructurar un escrito de protesta y elevarlo a las Cortes, si bien se decidió emplazar la reunión oficial para el viernes, conforme a sus estatutos27. Algunos ya hablaban de convocar una solemne manifestación de protesta el domingo en Madrid, jaleados por periódicos como La Iberia28. Los sucesos se precipitaron. La primera reunión del Círculo, de carácter tumultuario, se efectuó el jueves y, luego de programar una serie de medidas de carácter gremial, más o menos urgentes, propusieron el cierre del comercio madrileño, durante un día –el sábado siguiente– en señal de luto por la declaración oficial del cólera, pues partían de la consideración de su absoluta falsedad29.
EL MOTÍN DE LAS BANDERAS NEGRAS
El día anterior a la gran manifestación, el viernes 19 de julio de 1885, las tiendas de telas de la calle de Toledo, que en ese momento eran allí mayoritarias, aparecieron con los muestrarios y los dinteles de las puertas de sus comercios cubiertos con crespones, pañuelos o bandas de color negro. La panorámica desde la plaza Mayor era francamente fúnebre (Figura 7).
 A eso de las nueve y media de la mañana, las verduleras de la plaza de la Cebada, con unos cuantos chicuelos de poca edad, decidieron manifestarse contra la declaración del Gobierno. Mediante una suscripción de cinco céntimos por cabeza, a la que algunos caballeros allí presentes añadieron dos reales por barba, adquirieron la tela negra necesaria para hacer un estandarte. Lo tuvieron cortado y cosido en media hora, con un letrero en donde ponía: espárragos, lechugas y alcachofas contra el cólera. Del asta del mismo colgaron las mencionadas frutas. Todas se dotaron de un lazo negro, regalado por uno de los comerciantes, colocado sobre sus corpiños. Entretanto, a grandes voces, discutían si había motivos para fumigar las casas de los pobres y los puestos del mercado. Los guardias no conseguían restablecer la circulación y el Gobernador civil de Madrid envió un amplio retén para rodear en dos filas la plaza. Los gritos iban en aumento:
A eso de las nueve y media de la mañana, las verduleras de la plaza de la Cebada, con unos cuantos chicuelos de poca edad, decidieron manifestarse contra la declaración del Gobierno. Mediante una suscripción de cinco céntimos por cabeza, a la que algunos caballeros allí presentes añadieron dos reales por barba, adquirieron la tela negra necesaria para hacer un estandarte. Lo tuvieron cortado y cosido en media hora, con un letrero en donde ponía: espárragos, lechugas y alcachofas contra el cólera. Del asta del mismo colgaron las mencionadas frutas. Todas se dotaron de un lazo negro, regalado por uno de los comerciantes, colocado sobre sus corpiños. Entretanto, a grandes voces, discutían si había motivos para fumigar las casas de los pobres y los puestos del mercado. Los guardias no conseguían restablecer la circulación y el Gobernador civil de Madrid envió un amplio retén para rodear en dos filas la plaza. Los gritos iban en aumento:
¡No hay cólera, que hay hambre!
¡El cólera el de los pobres! ¡Qué traigan pan! ¡Abajo los del cólera!
Además del estandarte, aparecieron carteles con calaveras pintadas, en las cuales las tibias cruzadas eran sustituidas por espárragos y alcachofas; otros con féretros. Con grandes gritos, mediante los cuales proclamaban su intención de acercarse hasta las Cortes o la sede del Gobierno, se pusieron en marcha. En la plaza de San Millán las detuvo la policía, saludada con una lluvia de verduras de todas clases y algunas piedras.
 Otro grupo, desde la calle de Toledo se dirigió hacia la de Postas. Pasaron por la calle Imperial, frente al laboratorio municipal; se detuvieron y amenazaron con destruir los aparatos de desinfección y los equipos de fumigación. Lo impidieron los empleados del laboratorio y los guardias municipales. A continuación, intentaron hacer una barricada con los elementos de construcción destinados a la reparación de la calle Postas. El coronel Oliver y su ayudante, el señor Palma, al frente de un grupo de guardias municipales, intentó convencerlas de acabar con la manifestación. Recibieron una nueva lluvia de verduras y el citado militar una pedrada en el costado izquierdo; consiguió, pese a ello, hacerse con el fúnebre estandarte de la protesta, no sin ejercer violencia sobre la portadora, circunstancia propiciadora, probablemente, de la pedrada. Acabó llevándose el estandarte vencido a la sede del Gobierno civil, junto a dieciocho detenidos, trece hombres y cinco mujeres, mientras el grueso de los manifestantes se replegaba hacia la calle Toledo.
Otro grupo, desde la calle de Toledo se dirigió hacia la de Postas. Pasaron por la calle Imperial, frente al laboratorio municipal; se detuvieron y amenazaron con destruir los aparatos de desinfección y los equipos de fumigación. Lo impidieron los empleados del laboratorio y los guardias municipales. A continuación, intentaron hacer una barricada con los elementos de construcción destinados a la reparación de la calle Postas. El coronel Oliver y su ayudante, el señor Palma, al frente de un grupo de guardias municipales, intentó convencerlas de acabar con la manifestación. Recibieron una nueva lluvia de verduras y el citado militar una pedrada en el costado izquierdo; consiguió, pese a ello, hacerse con el fúnebre estandarte de la protesta, no sin ejercer violencia sobre la portadora, circunstancia propiciadora, probablemente, de la pedrada. Acabó llevándose el estandarte vencido a la sede del Gobierno civil, junto a dieciocho detenidos, trece hombres y cinco mujeres, mientras el grueso de los manifestantes se replegaba hacia la calle Toledo.
De la primitiva manifestación, un grupo de mujeres bajó por la calle de Ciudad Rodrigo; otro por la de la Pasa. Allí encontraron a un guardia que llevaba al Gobierno civil a dos muchachos, detenidos por vagancia e ir indocumentados. Las manifestantes exigieron su inmediata puesta en libertad, a lo que accedió el guardia, quien se puso a cubierto en cuanto pudo.
El Gobernador Civil, Raimundo Fernández Villaverde30, envió un retén de treinta guardias civiles a caballo para rodear el mercado de la cebada. Se mantuvieron allí hasta la una y media de la tarde. Cuando se retiraron, todo estaba en calma. En el ínterin, el Gobernador se presentó en la calle de Toledo, junto al Teniente de Alcalde del distrito de La Latina, Camilo Rodríguez. Fueron recibidos con grandes silbidos. Su despliegue de valor o de inconsciencia pudo acabar mal. Desde un edificio de la calle San Millán le tiraron un objeto que se destrozó a sus pies. Una verdulera, con mayor puntería, le lanzó una voluminosa lechuga y le manchó el cuello. El asunto se resolvió con los detenidos en el juzgado de guardia y la calle Toledo, sus aledañas y la plaza de la Cebada en paz31 (Figura 8).
La manifestación de los comerciantes: no existe el cólera en Madrid
El sábado 20 de junio aparecieron cerrados todos los comercios de la capital. Los que no lo hicieron por su gusto, se vieron presionados a hacerlo, como el ciudadano francés, afincado en Madrid, Capdeville, dueño del café francés. Con media puerta abierta permanecieron las boticas, carbonerías y tiendas de frutas.
En la calle de las Maldonadas, frente al mercado de la plaza de la Cebada, se estacionaron veinticuatro guardias civiles a caballo con su oficial al frente. Otra sección del mismo instituto en la plaza de San Millán. Alrededor de la plaza y por la calle Toledo se estableció un cordón de orden público, formado por cuatro agentes en cada esquina. También había fuerzas del orden cerca de los estancos y las tiendas de comestibles abiertas. En el interior de los mercados, parejas de guardias municipales. Frente a la fábrica de tabacos, parejas de la Guardia Civil y de la policía. Otros guardias civiles patrullaban a caballo, ante los rumores de manifestación por la tarde, cuando fuera el Rey a visitar a Nuestra Señora de Atocha. El Gobernador civil llamó a su presencia al Presidente del Círculo de la Unión Mercantil y le rogó la apertura de los comercios. Carlos Prast, le puso de manifiesto su incapacidad personal para hacerlo y, en contestación a los periodistas, les manifestó la mejor forma de acabar con el conflicto: que el Gobierno, a la mayor brevedad posible, declarara que no existe el cólera en Madrid.
La ciudad permaneció en paz, pero con miles de jóvenes dependientes con ganas de alboroto y muchas gentes enfadadas, por la declaración del cólera o por el cólera en sí mismo, dispuestas a cargar contra cualquier persona a quien consideraran responsable de la situación.
Por la tarde, los reyes salieron de palacio para visitar la basílica de Nuestra Señora de Atocha, según su costumbre sabatina. A la ida no pasó nada de particular, simplemente había mucha animación en la Puerta del Sol y en la carrera de San Jerónimo hasta las Cortes. A su paso, la muchedumbre se apartó respetuosa e incluso lanzó algún ¡viva!, si bien el tercer coche, en donde iba el Gobernador civil, fue retenido por la multitud, la cual le silbó, pero le permitió volver a pie al palacio de la Gobernación.
El regreso, a las siete de la tarde, tuvo otro cariz. La Puerta del Sol estaba abarrotada de gente. El primer coche, con don Alfonso XII y su esposa, fue vitoreado. En el segundo coche iba la Reina madre, Isabel II, y la Infanta doña Eulalia; a su paso se registraron algunos gritos contra la monarquía y algo cayó en el vehículo.
El Gobernador civil hizo fijar en la fachada del Ministerio de Gobernación un bando. En el mismo acusaba a los alborotadores de alterar el orden so pretexto de las medidas sanitarias. Por ello aconsejaba volver a sus casas a todas las personas de buena fe, pues estaba dispuesto a restablecerlo con toda firmeza. Fernández Villaverde, muy joven, se puso al frente de las operaciones. Como al salir con su coche le volvieran a silbar, se bajó del mismo y se enfrentó solo a los manifestantes, los cuales retrocedieron varias veces. El Gobernador tuvo el arrojo –algo inconsciente– de coger con sus manos a alguno de los amotinados y entregarlo a las fuerzas del orden (Figura 9).
 Cuando se habían marchado la casi totalidad de los manifestantes, la Guardia Civil cargó contra los grupos de alborotadores y curiosos todavía presentes. Realizaron las tres intimidaciones de corneta, según la ordenanza, y cargaron al trote. Los revoltosos huyeron y se reagruparon en las calles del Correo, Postas, Preciados, Carmen y Carretas. A partir de ese momento las cargas se sucedieron de seguido, con idéntico resultado: huida y reagrupamiento para seguir hostigando a los guardias civiles.
Cuando se habían marchado la casi totalidad de los manifestantes, la Guardia Civil cargó contra los grupos de alborotadores y curiosos todavía presentes. Realizaron las tres intimidaciones de corneta, según la ordenanza, y cargaron al trote. Los revoltosos huyeron y se reagruparon en las calles del Correo, Postas, Preciados, Carmen y Carretas. A partir de ese momento las cargas se sucedieron de seguido, con idéntico resultado: huida y reagrupamiento para seguir hostigando a los guardias civiles.
Tras el ballet revolucionario, desarrollado en clave chusca, con jóvenes apaleados, señoras con ataques de nervios, jovencitos agazapados en la fuente que entonces había en la puerta del Sol, huyendo empapados, o un niño cruzando tranquilamente la plaza de la mano de su abuelo, llegó la tragedia. Los manifestantes iban provistos de armas cortas y de ellos partieron los primeros tiros. Además de los insultos y las piedras, las fuerzas del orden empezaron a recibir disparos. Los mismos fueron contestados por la Guardia Civil, de a caballo, en grupos de cuatro en cuatro, disparando sus fusiles al aire. A las diez de la noche llegó a la Puerta del Sol el ministro de la Guerra vestido de paisano. Habló con los jefes de los distintos grupos y dio órdenes para que la Guardia Civil fuera sustituida por soldados del batallón de Puerto Rico.
 A partir de ese momento se fueron distendiendo las posturas; parece que incluso hubo intercambio de cigarrillos entre los jóvenes y los soldados. La pacificación llegó tras la tragedia. A las dos de la madrugada había acabado todo, si bien la totalidad de los disparos no pudieron hacerse al aire pues hubo dos jóvenes muertos. Uno de un disparo en el corazón y otro con un balazo en la cabeza32.
A partir de ese momento se fueron distendiendo las posturas; parece que incluso hubo intercambio de cigarrillos entre los jóvenes y los soldados. La pacificación llegó tras la tragedia. A las dos de la madrugada había acabado todo, si bien la totalidad de los disparos no pudieron hacerse al aire pues hubo dos jóvenes muertos. Uno de un disparo en el corazón y otro con un balazo en la cabeza32.
El asunto acabó con la visita a palacio de los representantes del Círculo de la Unión Mercantil, para presentar al monarca sus respetos y reivindicaciones, aunque con la desmesurada petición del no reconocimiento de una enfermedad existente causaron, aunque fuera indirectamente, el motín y la muerte de dos jóvenes obreros, sucesos duramente criticados por los representantes del Círculo ante S.M., junto al desmarque de los periódicos de izquierda revolucionaria al advertir que esa no era su batalla, sino la de los comerciantes, a quienes consideraban sus explotadores33.
En cuanto a la derogación de la declaración de la epidemia, el monarca, sabia y prudentemente, luego de dejar claras sus limitaciones como Rey constitucional, les puso de manifiesto sus condolencias por las pérdidas causadas, si bien, les manifestó de manera adusta, solo incumbe a la divina Providencia derogarla, haciendo cesar la epidemia que por fortuna hasta ahora causa pocas víctimas34 (Figura 10).





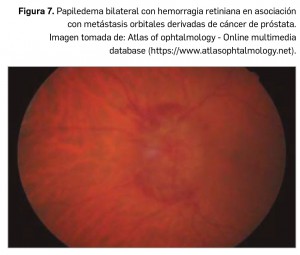
 El nervio óptico abandona la órbita a través del foramen óptico, cerca de la arteria oftálmica y transcurre hacia atrás hasta unirse al nervio contralateral en el quiasma óptico, donde se producirá una decusación parcial: los axones de las células ganglionares del lado nasal de la retina pasan al lado opuesto. El tracto óptico, formado por fibras nasales contralaterales y temporales homolaterales, abandona el quiasma y se extiende hasta el cuerpo geniculado lateral, aunque algunas fibras abandonan el tracto antes de llegar al cuerpo geniculado y pasan al colículo superior, encargándose del reflejo pupilar. Los axones de los cuerpos celulares del cuerpo geniculado lateral forman la radiación óptica, la cual entrará en el hemisferio en la parte más posterior de la cápsula interna. Transcurren por las profundidades de los lóbulos parietal y temporal y terminan en la corteza calcarina del lóbulo occipital.
El nervio óptico abandona la órbita a través del foramen óptico, cerca de la arteria oftálmica y transcurre hacia atrás hasta unirse al nervio contralateral en el quiasma óptico, donde se producirá una decusación parcial: los axones de las células ganglionares del lado nasal de la retina pasan al lado opuesto. El tracto óptico, formado por fibras nasales contralaterales y temporales homolaterales, abandona el quiasma y se extiende hasta el cuerpo geniculado lateral, aunque algunas fibras abandonan el tracto antes de llegar al cuerpo geniculado y pasan al colículo superior, encargándose del reflejo pupilar. Los axones de los cuerpos celulares del cuerpo geniculado lateral forman la radiación óptica, la cual entrará en el hemisferio en la parte más posterior de la cápsula interna. Transcurren por las profundidades de los lóbulos parietal y temporal y terminan en la corteza calcarina del lóbulo occipital.
 La mayoría de pacientes diagnosticados en fase inicial y con tratamiento adecuado presentan una evolución favorable. Los tratamientos van encaminados a mejorar la irrigación arterial de la papila óptica disminuyendo la presión intraocular y aumentando el flujo sanguíneo. Entre ellos, los bloqueadores beta son el tratamiento de primera elección, siempre que no haya contraindicaciones para su uso; actúan disminuyendo la secreción del humor acuoso, pero pueden producir broncoespasmo, bradicardia e hipotensión. Entre ellos se encuentra el betaxolol que puede emplearse en broncópatas.
La mayoría de pacientes diagnosticados en fase inicial y con tratamiento adecuado presentan una evolución favorable. Los tratamientos van encaminados a mejorar la irrigación arterial de la papila óptica disminuyendo la presión intraocular y aumentando el flujo sanguíneo. Entre ellos, los bloqueadores beta son el tratamiento de primera elección, siempre que no haya contraindicaciones para su uso; actúan disminuyendo la secreción del humor acuoso, pero pueden producir broncoespasmo, bradicardia e hipotensión. Entre ellos se encuentra el betaxolol que puede emplearse en broncópatas.
 El paciente presenta una pérdida indolora y aguda del campo visual, que suele ser percibida por la mañana al despertar, ya que la disregulación hemodinámica podría aumentar por la noche con la caída de la tensión arterial. El diagnóstico inicial es de presunción y no se confirma hasta transcurridos dos meses, cuando se ha resuelto el edema del disco óptico y ha aparecido una palidez difusa o sectorial del nervio óptico con estrechamiento arteriolar. El pronóstico visual es malo, pues el riesgo de bilateralización es aproximadamente de un 15%, apareciendo entonces una imagen de edema de nervio óptico con atrofia óptica contralateral (pseudo Foster-Kennedy).
El paciente presenta una pérdida indolora y aguda del campo visual, que suele ser percibida por la mañana al despertar, ya que la disregulación hemodinámica podría aumentar por la noche con la caída de la tensión arterial. El diagnóstico inicial es de presunción y no se confirma hasta transcurridos dos meses, cuando se ha resuelto el edema del disco óptico y ha aparecido una palidez difusa o sectorial del nervio óptico con estrechamiento arteriolar. El pronóstico visual es malo, pues el riesgo de bilateralización es aproximadamente de un 15%, apareciendo entonces una imagen de edema de nervio óptico con atrofia óptica contralateral (pseudo Foster-Kennedy).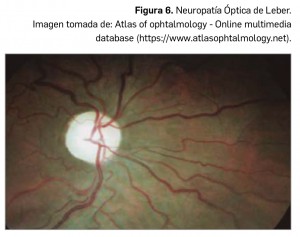 En estos pacientes se recomienda realizar electrocardiograma para descartar trastornos de la conducción cardiaca como el Síndrome de Wolff-Parkinson-White o el síndrome de Lown-Ganon-Levine. Ante la sospecha de neuropatía óptica de Leber, se debe realizar un estudio genético ya que, según la mutación encontrada, el pronóstico varía: aunque en general es malo, se ha descrito mejoría en el 60% de los pacientes con mutaciones en las posiciones 3460 y 14484 del ADN mitocondrial, y en el 5% en la 11778.
En estos pacientes se recomienda realizar electrocardiograma para descartar trastornos de la conducción cardiaca como el Síndrome de Wolff-Parkinson-White o el síndrome de Lown-Ganon-Levine. Ante la sospecha de neuropatía óptica de Leber, se debe realizar un estudio genético ya que, según la mutación encontrada, el pronóstico varía: aunque en general es malo, se ha descrito mejoría en el 60% de los pacientes con mutaciones en las posiciones 3460 y 14484 del ADN mitocondrial, y en el 5% en la 11778.