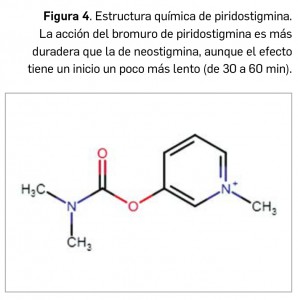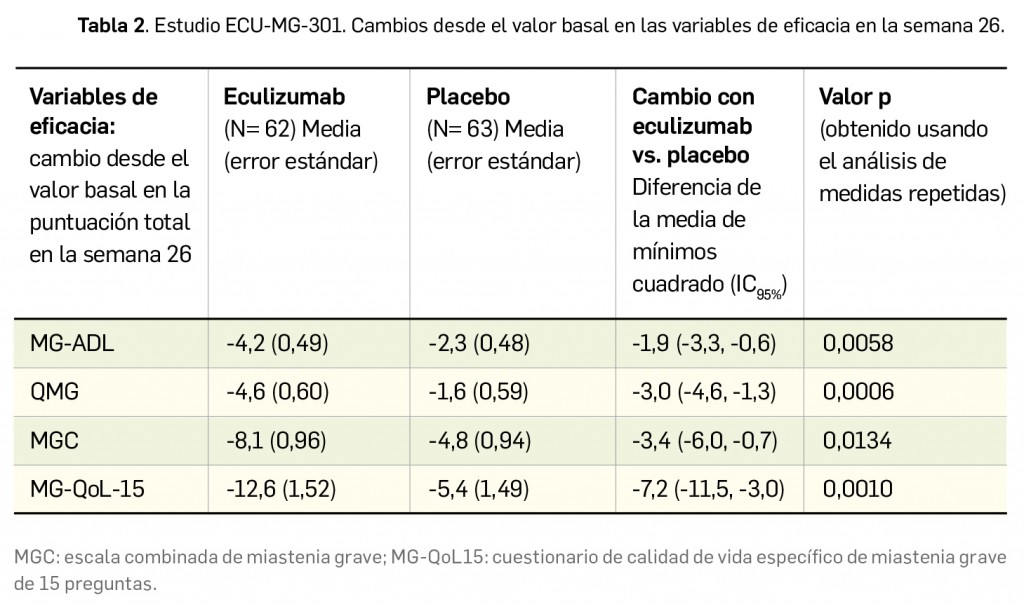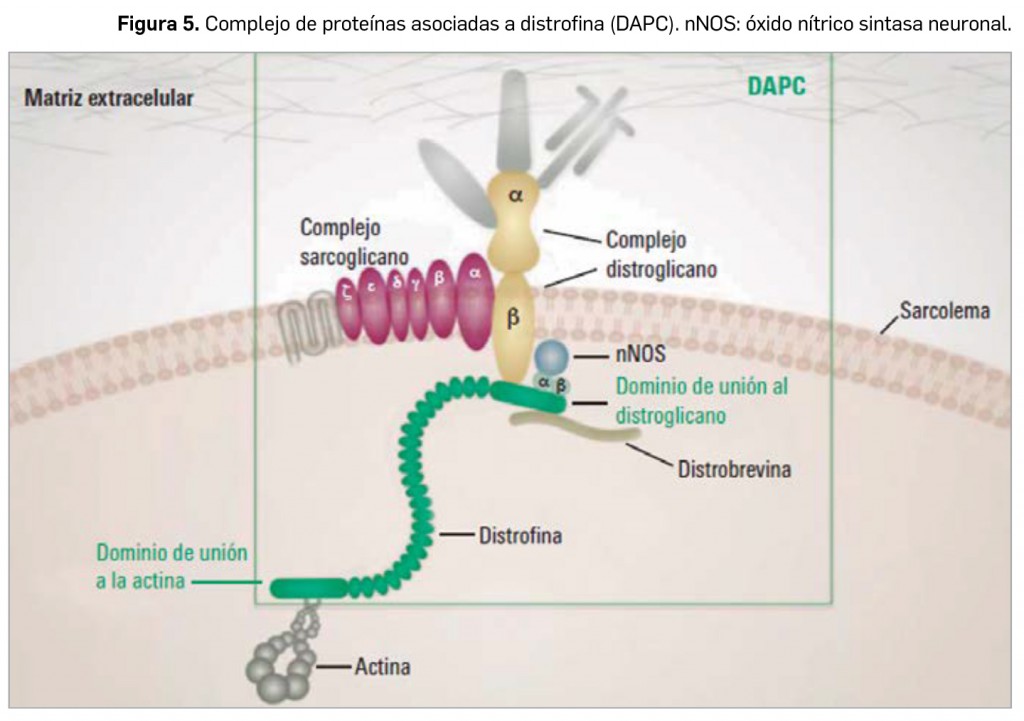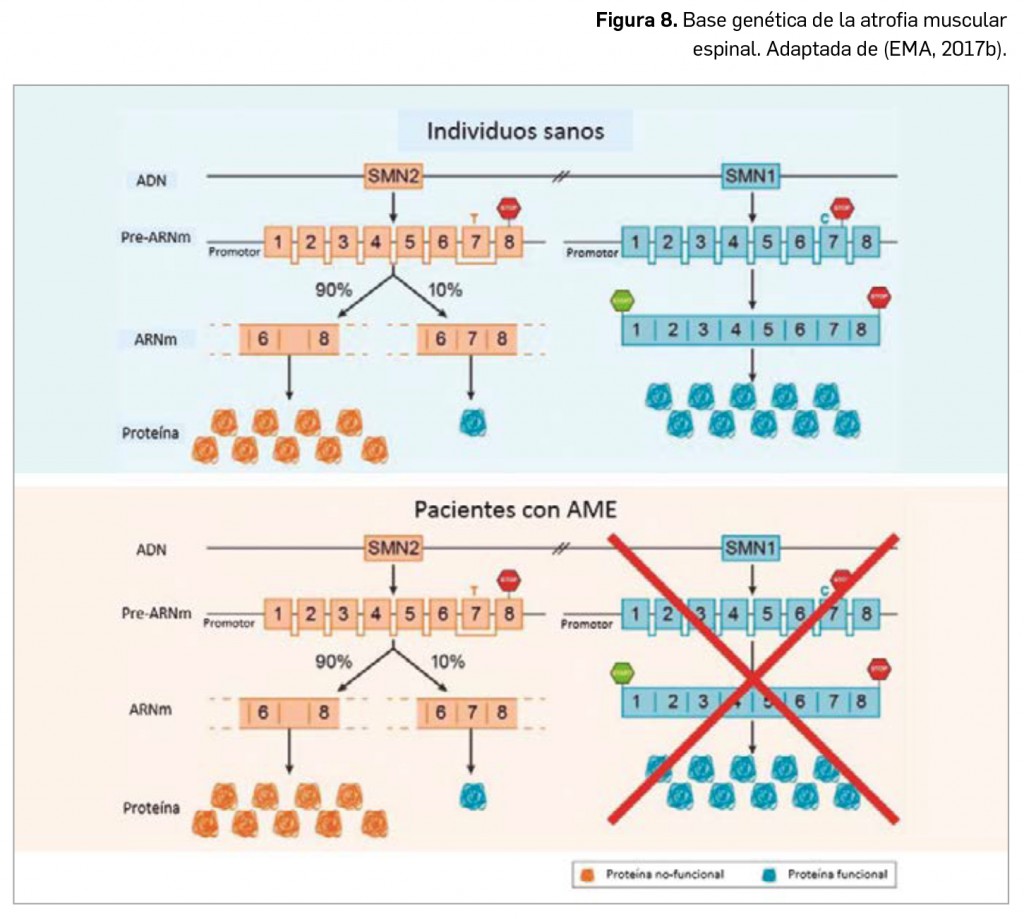
CÓMO LOCALIZAR CAMBIOS DE NOMBRE Y DE LABORATORIO CON BOT PLUS
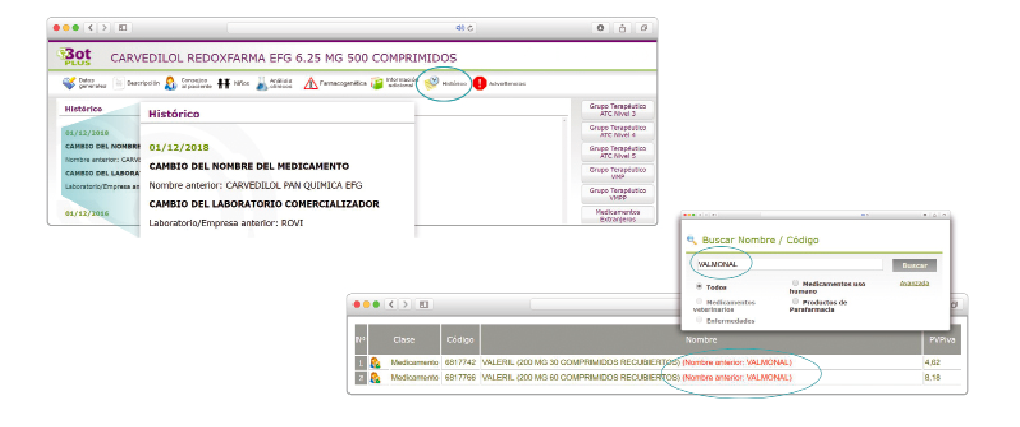
Además de la información que se incluye en los listados mensuales publicados en PAM, en BOT PLUS se incluye un apartado de Histórico, en las fichas de medicamentos, en el que se presenta información referente a cambios que haya sufrido anteriormente el medicamento o producto, entre otros, los cambios de nombre y los cambios de laboratorio. Esta información también está disponible para productos sanitarios financiados o dietoterápicos.
Se añade la posibilidad de visualización de las situaciones anteriores (o incluso futuras) relacionadas con un cambio de nombre.
Con automatismos que nos permiten localizar un medicamento que haya cambiado de nombre, independientemente de cuál usemos.
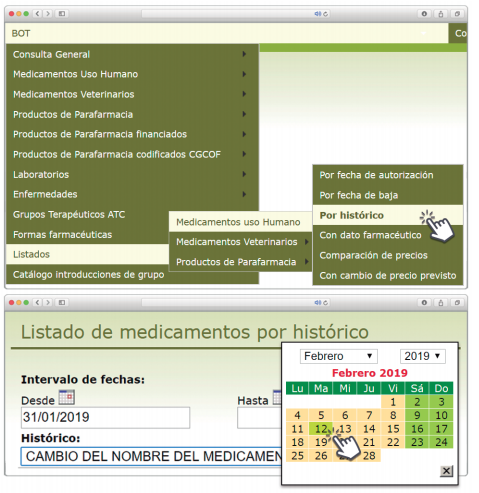
Posibilidad de generar listados por Histórico
Además de la información existente en Histórico, se permite la explotación de la información incluida en BOT PLUS en este apartado, mediante la integración de la información almacenada en Histórico en el apartado de Listados de BOT PLUS, que permite realizar consultas entre rangos de fechas y por un concepto en concreto de entre los almacenados en el apartado de Histórico. Entre ellos se incluyen, precisamente, los conceptos “Cambio del nombre del medicamento” y “Cambio del laboratorio comercializador”.