Resumen
Es habitual en la farmacia comunitaria encontrarnos con pacientes que presentan ciertas dificultades a la hora de ingerir comprimidos o cápsulas. BOT PLUS identifica, a través de unos pictogramas concretos, a aquellos medicamentos que pueden ser manipulados por el paciente para facilitar esta deglución. De esta manera, y de forma rápida y visual, el farmacéutico podrá conocer las acciones que puedan llevarse a cabo para facilitar la administración del medicamento.
Siguiendo con la revisión de las novedades incorporadas en la versión 2020 de BOT PLUS, en esta ocasión nos vamos a centrar en la información que se ha incluido para poder identificar las formas farmacéuticas sólidas que se administran por vía oral que se pueden manipular para facilitar su deglución en aquellos pacientes que presenten dificultades a la hora de ingerirlas.
Cuando un paciente presenta dificultades a la hora de ingerir un medicamento (estos problemas son especialmente comunes en niños, en ancianos o en pacientes con ciertas patologías como Parkinson, ictus o con problemas de disfagia), en primer lugar el médico puede seleccionar una forma farmacéutica que facilite esta deglución, como serían formas farmacéuticas líquidas (jarabes o soluciones orales), efervescentes (tanto en granulado como en comprimidos), comprimidos masticables, dispersables o bucodispersables.
Pero en muchas ocasiones el principio activo seleccionado solo está disponible en medicamentos formulados como comprimidos o cápsulas. Estas formas galénicas, en algunas ocasiones (no siempre es posible), pueden manipularse partiendo los comprimidos o abriendo las cápsulas. Por ello, a partir de ahora, BOT PLUS identificará los medicamentos cuyos comprimidos puedan partirse o disolverse, o cuyas cápsulas puedan abrirse, a través de unos pictogramas concretos, que de forma rápida y visual indicarán al farmacéutico las acciones que puedan llevarse a cabo para facilitar la administración del medicamento.
En la ficha de cada medicamento en BOT PLUS se ha incluido toda esta información en forma de mensajes de advertencia y pictogramas:
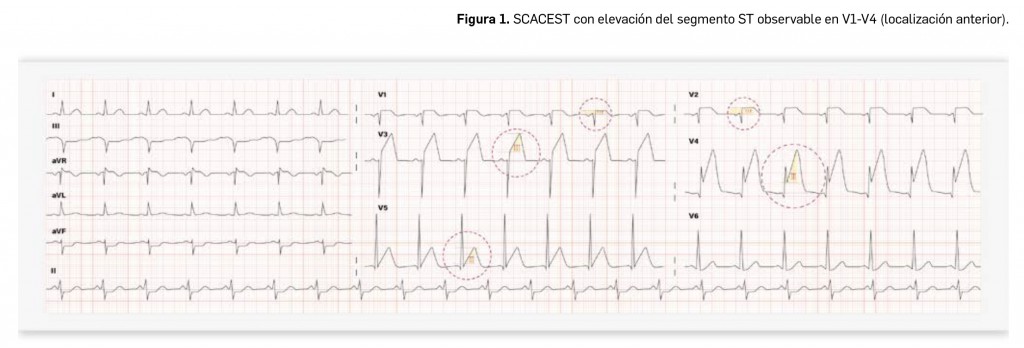
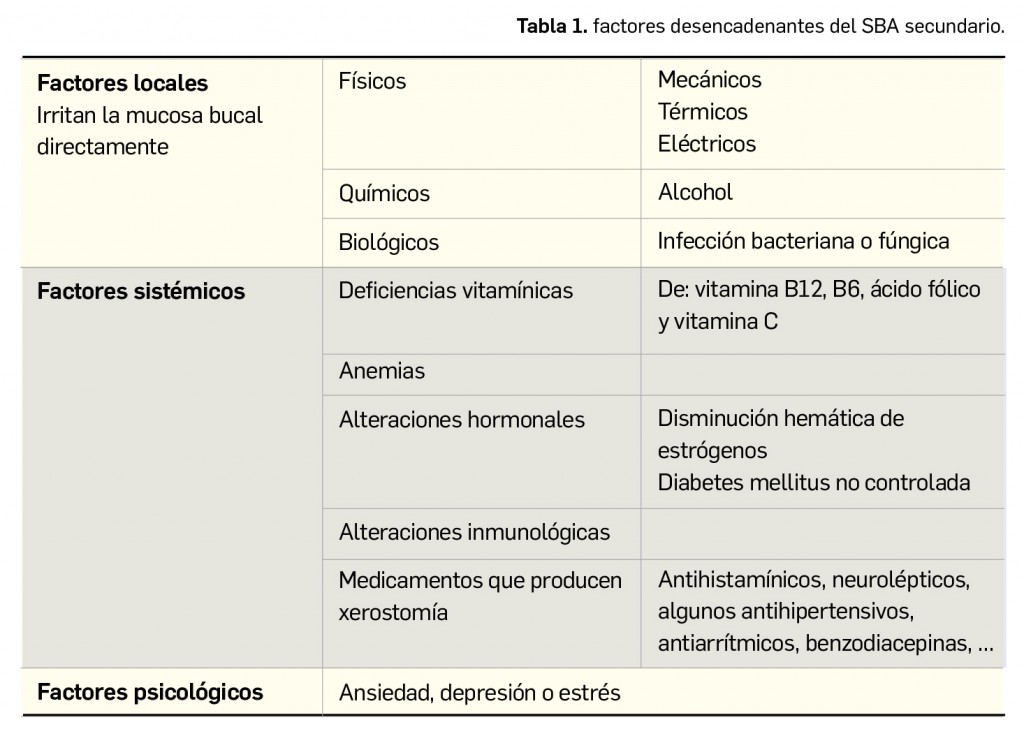
- Aquellas presentaciones que se puedan manipular para facilitar la deglución llevan asignadas un mensaje de advertencia que indica “DEGLUCION: ADAPTADO PARA PACIENTES CON DIFICULTADES DE DEGLUCION” asociado a un pictograma verde (Figura 1) con un comprimido partido para indicarnos que el medicamento se puede partir (en el caso de los comprimidos) o abrir (para las cápsulas).
Junto a esta información general, se incluyen mensajes secundarios que especifican la manipulación recogida en ficha técnica, como la posibilidad de partir, masticar o machacar un comprimido, la posibilidad de disgregarlo en líquido o, en el caso de cápsulas, la posibilidad de dispersar su contenido en líquidos o alimentos blandos, entre otras. Igualmente, y en caso de que fuera posible, se recoge en este punto si el medicamento puede suspenderse en líquido y administrarlo a través de sondas nasogástricas.
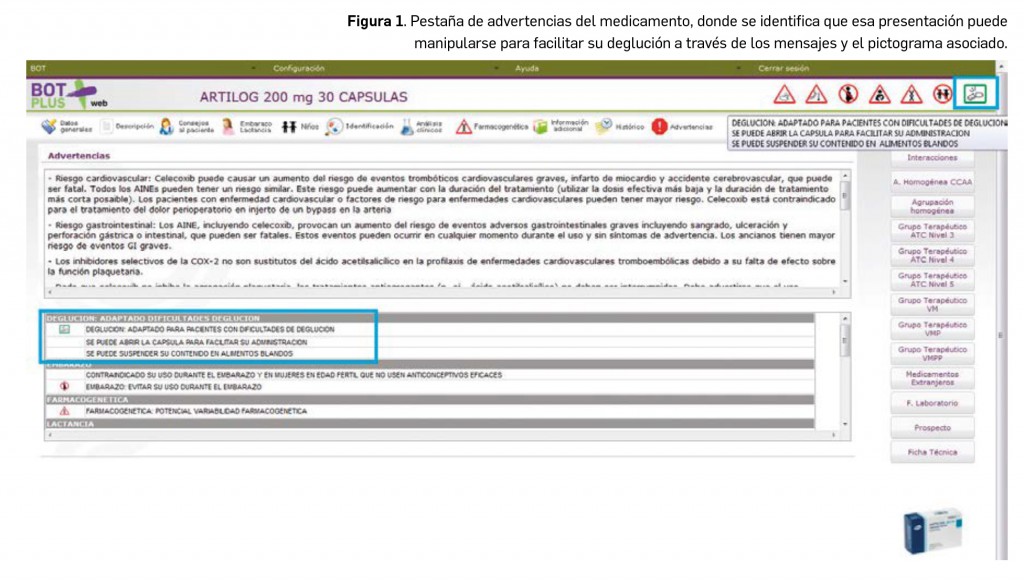
- En cambio, las presentaciones que no pueden manipularse para facilitar la deglución llevan asignadas un mensaje de advertencia que indica “DEGLUCION: NO ADAPTADO PARA PACIENTES CON DIFICULTADES DE DEGLUCION” asociado a un pictograma rojo de prohibición (Figura 2) para indicarnos que el comprimido o la cápsula debe tragarse entero.
Si no aparece ningún pictograma, será una forma farmacéutica que ya de por sí facilita la deglución del medicamento.
Además, la codificación de esta información, permite buscar medicamentos en base a estos criterios a través de las búsquedas libres. Para ello, debemos seleccionar en el apartado “Advertencias” las opciones de “adaptado o no adaptado para pacientes con difcultades de deglución” y seleccionar también el resto de condiciones de búsqueda que nos interesen (por ejemplo, un principio activo concreto) (Figura 3).
La información relativa a la posibilidad de manipular estas formas farmacéuticas sólidas, que se encuentra codificada en BOT PLUS en la pestaña de Advertencias, se ha incluido en base a la información disponible en las fichas técnicas de estos medicamentos. Para ello, se ha revisado la información de más de 15.000 medicamentos de uso humano, de los cuales aproximadamente 5.000 pueden manipularse y 10.000 no.
La posibilidad de encontrar esta información en cada ficha de los medicamentos supone una importante ayuda para el farmacéutico, con el objetivo de poder dar indicaciones a los pacientes sobre la manipulación de una presentación concreta. Además, la codificación de esta información en BOT PLUS, unido a la posibilidad de incluir la composición exacta del medicamento u otras condiciones de búsqueda a través de las búsquedas libres, puede resultar útil al farmacéutico en la sustitución de medicamentos en pacientes con problemas de deglución, en los que interese buscar medicamentos que puedan manipularse. No obstante, siempre se aconseja consultar los listados oficiales respecto a la sustitución.