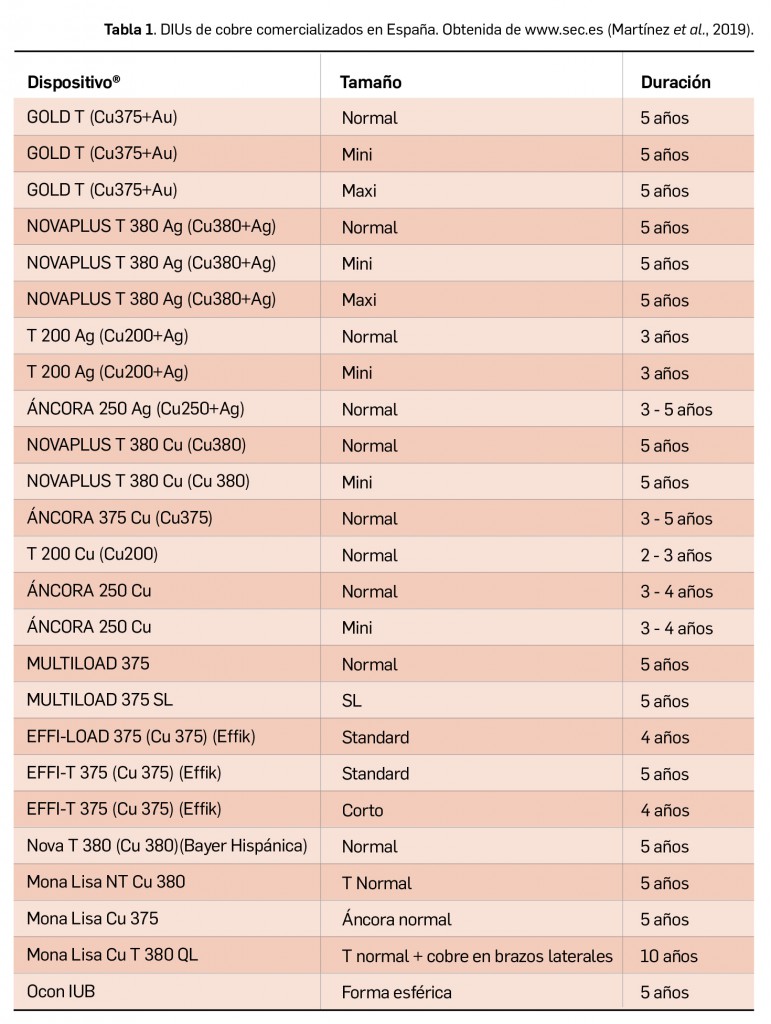
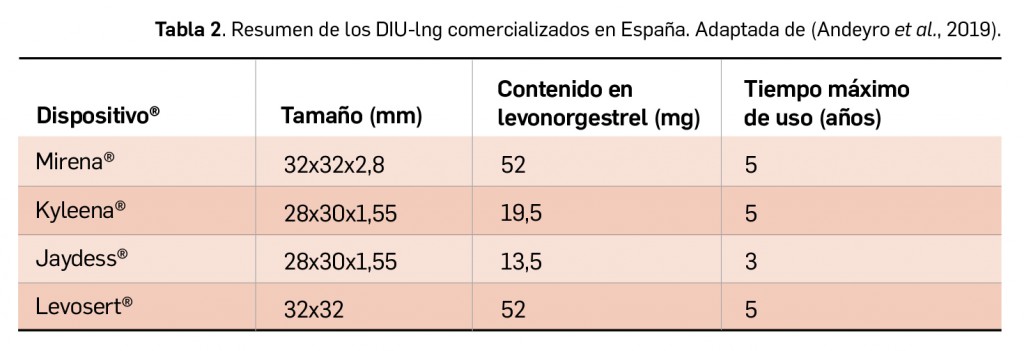
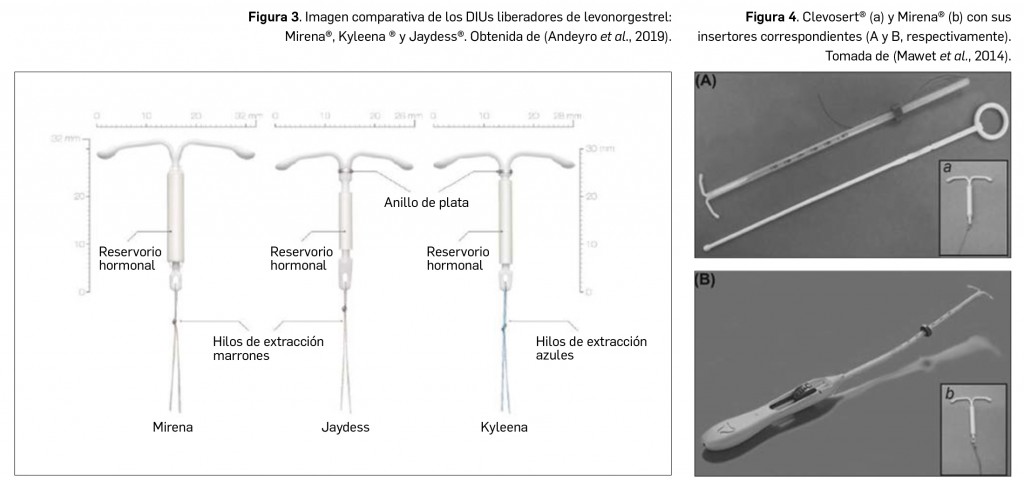
Resumen
El Consejo General de Colegios Oficiales de Farmacéuticos a través de su base de datos BOT PLUS facilita la labor asistencial del farmacéutico adaptándose a sus necesidades. A través de la codificación de la composición de los medicamentos autorizados por la AEMPS y de la composición de los productos de parafarmacia, el farmacéutico puede buscar un medicamento o producto con una composición concreta o identificar aquellos que se deben evitar en pacientes con ciertas alergias o intolerancias alimenticias.
BOT PLUS tiene codificada la composición cualitativa y cuantitativa de los medicamentos autorizados por la AEMPS, incluyendo en la misma tanto los principios activos como los excipientes de declaración obligatoria recogidos en la Circular 1/2018 de la AEMPS. Todas estas informaciones relativas a la composición proceden de las fichas técnicas autorizadas por la AEMPS para cada medicamento.
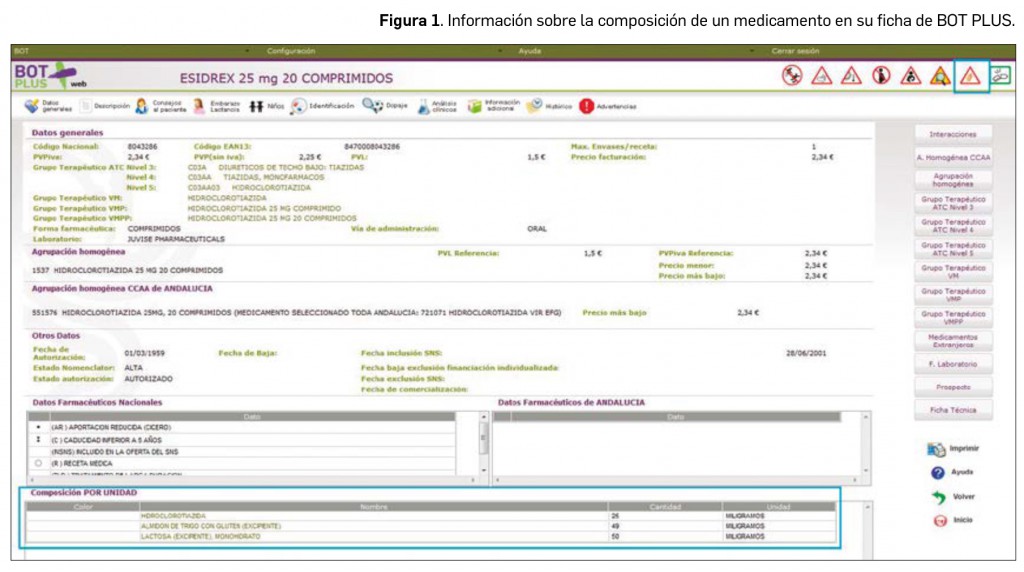
Al acceder a la ficha de un medicamento, se puede comprobar en la pantalla inicial si contiene excipientes de declaración obligatoria. En muchos casos, se ha incluido, además, una advertencia en los apartados de Consejos al paciente y Advertencias para aquellos medicamentos que deberían ser utilizados con precaución por pacientes que presenten alergias o intolerancias a estos excipientes (Figura 1). En algunos medicamentos, se puede visualizar, incluso, un pictograma específico en la parte superior derecha de su ficha (por ejemplo, pictograma de advertencia de uso en pacientes celiacos por el contenido en gluten).
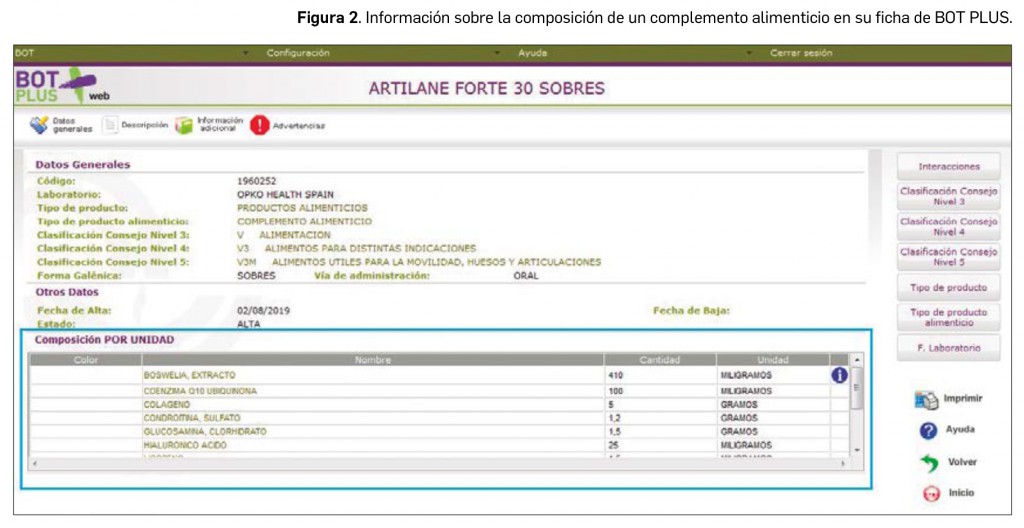
En el caso de los productos de parafarmacia, se está avanzando actualmente en la codificación de la composición de los cosméticos, complementos alimenticios, productos sanitarios y biocidas. Por ejemplo, en la ficha de los complementos alimenticios, podemos observar la composición de los nutrientes o de sustancias con un efecto nutricional o fisiológico (Figura 2).
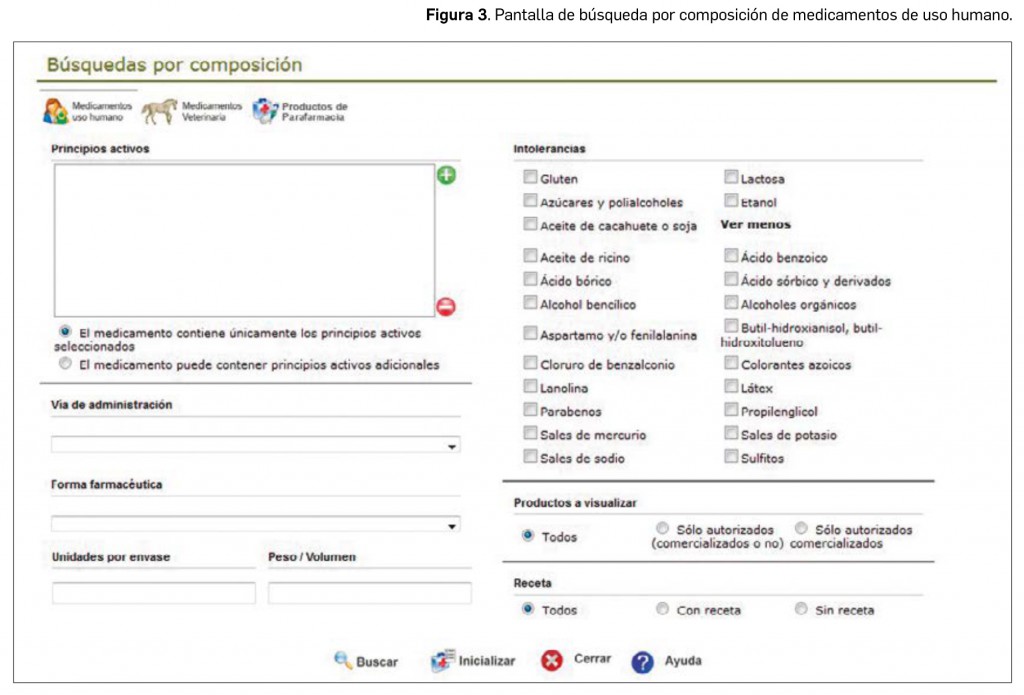
No obstante, con el objetivo de hacer más accesible la información de BOT PLUS y facilitar la labor asistencial de los farmacéuticos, este año se ha modificado el buscador por composición (Figura 3). Este buscador permite, entre otras funciones, filtrar los diferentes medicamentos o productos de parafarmacia según su principio activo o ingrediente, forma farmacéutica, número de unidades y dosificación. Esta búsqueda resulta especialmente útil en el caso de medicamentos o productos con más de un principio activo o ingrediente y en aquellos pacientes que presentan alergias o intolerancias alimenticias. A dicho buscador puede accederse desde la parte derecha de la pantalla inicial o a través del menú BOT / Consulta General /Búsqueda por composición.
De forma similar a lo que ocurre con otros tipos de búsquedas de la aplicación, las búsquedas por composición se dividen en bloques que hacen referencia al tipo de registros que se van a mostrar en el listado de resultados:
- Medicamentos de uso humano. A través de esta pantalla se generará un listado de medicamentos con uno o más principios activos concretos (se puede incluir también la sal y/o cantidades concretas) y/o uno o varios excipientes de declaración obligatoria, o sin estos últimos. Asimismo, se pueden incluir otras condiciones de búsqueda: forma farmacéutica, vía de administración, unidades del envase o peso/volumen. Por último, también se puede incluir el estado de autorización y sus condiciones de dispensación en cuanto a la necesidad o no de receta médica para la dispensación de esos medicamentos.
- Medicamentos veterinarios. Se obtendrá un listado de medicamentos veterinarios con uno o más principios activos concretos, pudiendo elegir si los medicamentos buscados deben incluir solo esos principios activos o pueden incluir otros fármacos adicionales. De forma similar a la opción anterior, se pueden incluir otras condiciones de búsqueda: forma farmacéutica, vía de administración, unidades del envase o peso/volumen. Por último, también se puede añadir el estado de autorización y sus condiciones de dispensación en cuanto a la necesidad o no de receta médica para la dispensación de esos medicamentos.
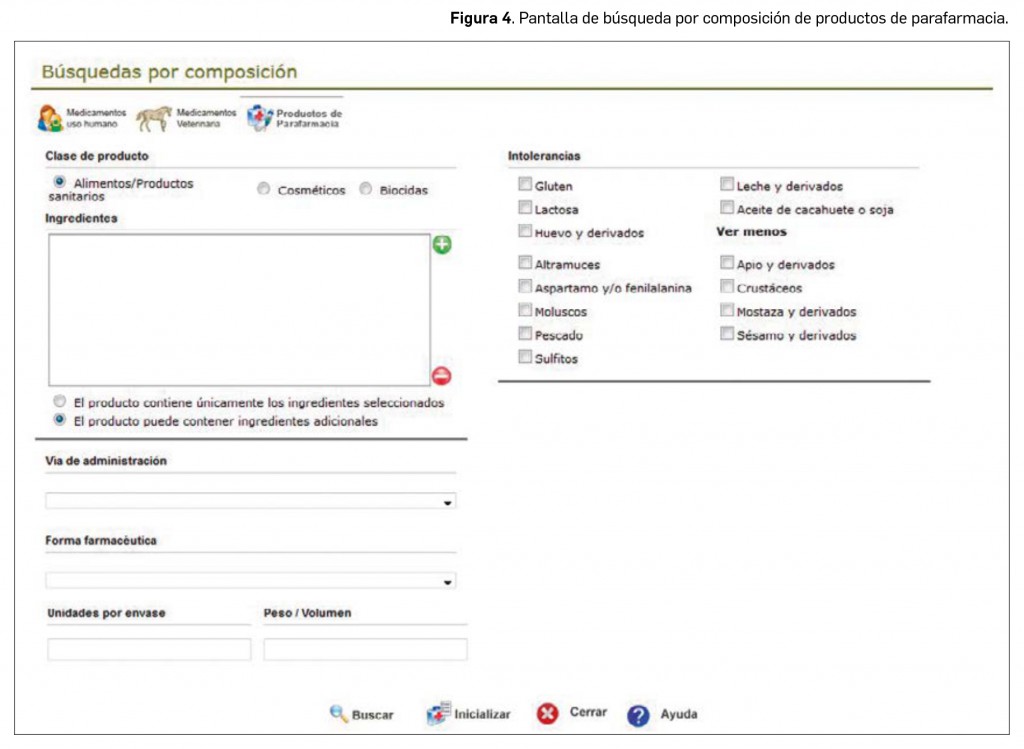
- Productos de parafarmacia. En esta opción (Figura 4) se debe definir primero qué clase de productos se quiere buscar: alimentos/productos sanitarios, cosméticos o biocidas. Posteriormente, se deben agregar como condición de búsqueda los ingredientes que se están buscando (si deben contener solo estos ingredientes o los productos buscados pueden contener ingredientes adicionales), pudiendo también excluir de la búsqueda productos con ciertos ingredientes que pueden causar alergias o intolerancias en el paciente. Finalmente, se pueden añadir otras condiciones de búsqueda: forma farmacéutica, vía de administración, unidades del envase o peso/volumen.
A modo de conclusión, hay que destacar que, mediante la codificación de la composición de los medicamentos autorizados por la AEMPS –tanto de sus principios activos como de sus excipientes de declaración obligatoria–, y la codificación de los ingredientes de los productos de parafarmacia en BOT PLUS, el farmacéutico comunitario puede buscar medicamentos o productos con unos principios activos o ingredientes concretos, e identificar aquellos que deben usarse con precaución en pacientes con ciertas alergias o intolerancias alimenticias. Todo ello en aras de facilitar su labor asistencial.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}