El Comité Europeo para la Evaluación de Riesgos en Farmacovigilancia (PRAC) ha acordado cambios en la información autorizada de las fichas técnicas y de los prospectos de los medicamentos europeos por motivos de seguridad.
Una vez que se revisan y evalúan los datos de los informes periódicos de seguridad (IPS; en inglés PSUR), de forma colaboradora entre las 27 agencias nacionales, se presentan los cambios y se acuerdan en las reuniones mensuales del PRAC. A continuación se muestran los últimos cambios de información de seguridad acordados recientemente en el PRAC.
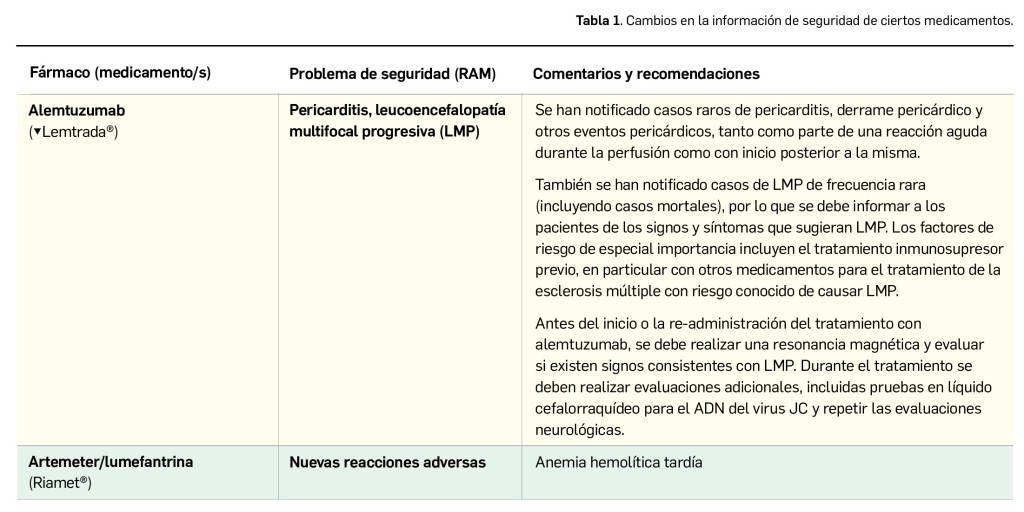
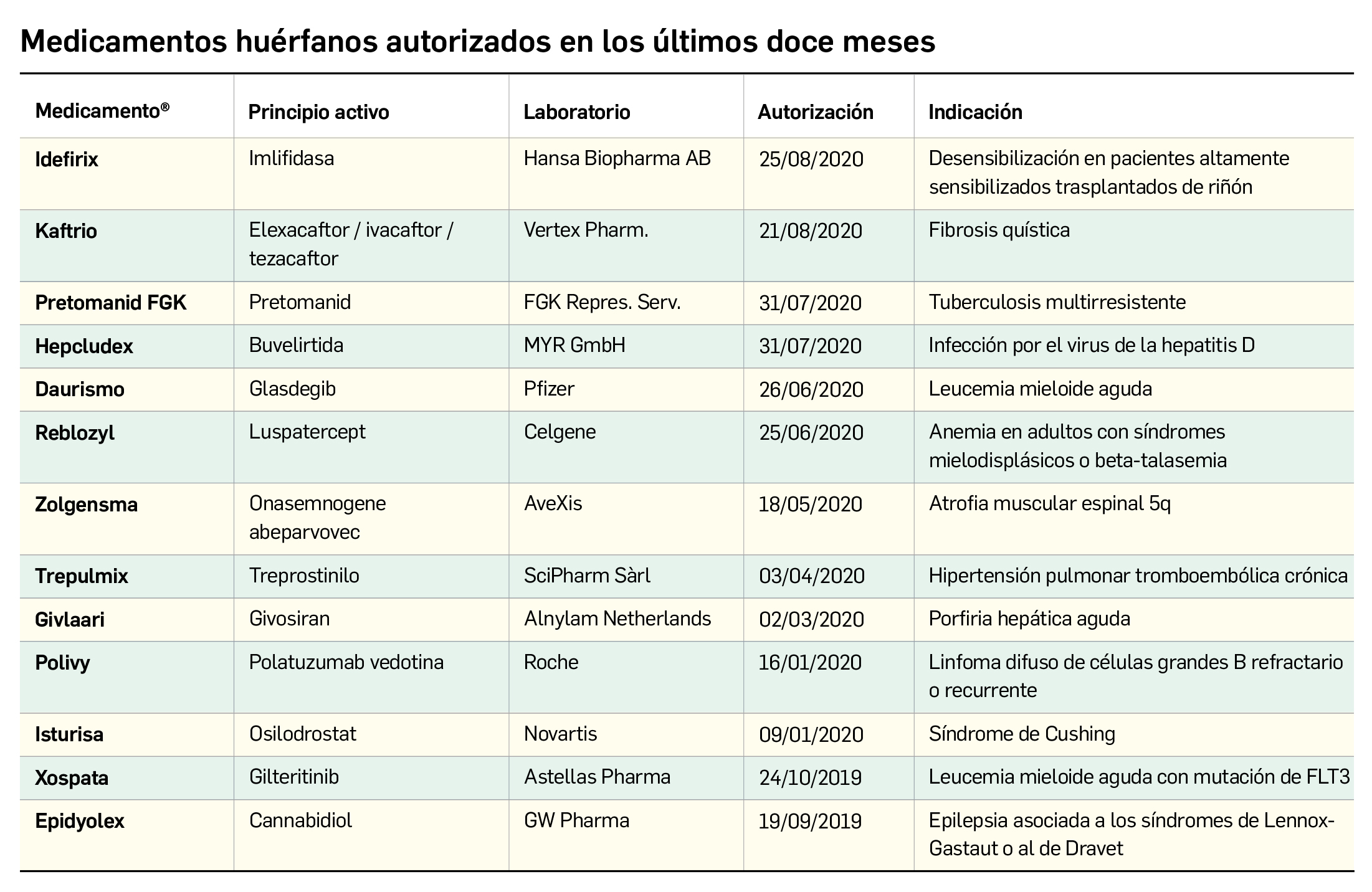
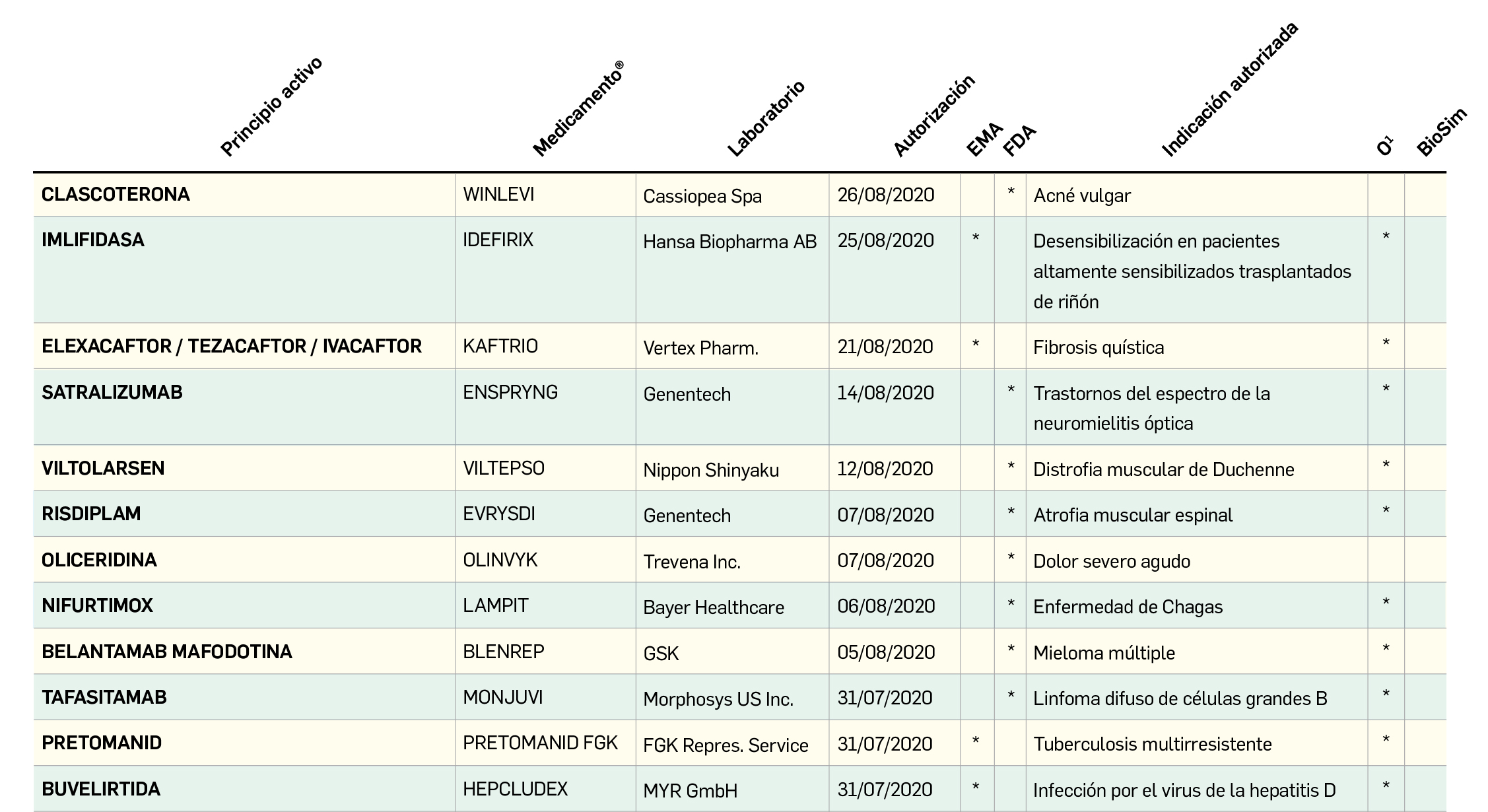
El Comité europeo para la Evaluación de Riesgos en Farmacovigilancia (PRAC) ha acordado cambios en las fichas técnicas y los prospectos de los siguientes medicamentos, siendo los más importantes los que se describen en la Tabla 1, según informa la AEMPS en sus Boletines Mensuales de abril y mayo de 2020.
Las fichas técnicas y prospectos de los medicamentos pueden consultarse en la web de la AEMPS, dentro de la sección CIMA: Centro de Información Online de Medicamentos.
Referencias
- Agencia Española de Medicamentos y Productos Sanitarios. Nueva información de seguridad procedente de la evaluación periódica de los datos de farmacovigilancia. Boletín Mensual de la AEMPS sobre Medicamentos de uso Humano, Abril 2020. Publicado el 30 de junio de 2020, páginas 9 a 12. Disponible en: https://www.aemps.gob.es/informa/boletines-AEMPS/boletinMensual/2020/boletin-mensual-MUH_abril-2020.pdf?x21576 (consultado 03 de septiembre de 2020).
- Agencia Española de Medicamentos y Productos Sanitarios. Nueva información de seguridad procedente de la evaluación periódica de los datos de farmacovigilancia. Boletín Mensual de la AEMPS sobre Medicamentos de uso Humano, Mayo 2020. Publicado el 28 de julio de 2020, páginas 13 a 16. Disponible en: https://www.aemps.gob.es/informa/boletines-AEMPS/boletinMensual/2020/boletin-mensual-MUH_mayo-2020.pdf?x21576 (consultado 03 de septiembre de 2020).
Información importante
El Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano (SEFV-H) se basa en el programa de notificación espontánea de un profesional sanitario (médico, odontólogo, farmacéutico, enfermero, otros) o de un ciudadano, de una sospecha de relación entre un medicamento (incluidos vacunas, sueros, gases medicinales, fórmulas magistrales, plantas medicinales) y un síntoma o signo adverso (reacción adversa, RAM) que manifieste el paciente o familiar (programa de tarjeta amarilla). El Real Decreto 577/2013 de Farmacovigilancia de medicamentos de uso humano (BOE núm. 179, de 27 de julio de 2013) entró en vigor el 28 de julio de 2013. La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) coordina el SEFV-H. A su vez se integra en el Sistema Europeo de Farmacovigilancia que desde 1995 coordina la Agencia Europea de Medicamentos (EMA), y participa desde 1984 en el Programa Internacional de Farmacovigilancia de la OMS, junto con más de 130 países.
¿Qué notificar? Se deben notificar las sospechas de RAM:
Con medicamentos autorizados, incluidas las de aquellos que se hayan utilizado en condiciones diferentes a las autorizadas o con medicamentos extranjeros importados con autorización de la AEMPS.
Principalmente las RAM ‘graves’ (mortales, o que amenacen la vida, prolonguen o provoquen una hospitalización, causen incapacidad o sean médicamente importantes y las trasmisiones de un agente infeccioso a través de un medicamento) o RAM ‘inesperadas’ de cualquier medicamento
Con medicamentos de ‘seguimiento adicional’ (durante sus primeros 5 años desde la autorización, identificados con un triángulo negro invertido (▼) a la izquierda del nombre del medicamento en el material informativo, en el prospecto y en la ficha técnica); ver la lista mensual de los medicamentos con “triángulo negro” en la web de la AEMPS: https://www.aemps.gob.es/vigilancia/medicamentosUsoHumano/seguimiento_adicional.htm#lista_europea
Las que sean consecuencia de ‘errores de medicación’, que ocasionen daño en el paciente,
Las originadas por ‘interacciones’ con medicamentos, plantas medicinales, incluso alimentos (zumo de pomelo, ahumados, crucíferas, etc).
¿Cómo notificar?
No olvide notificar cualquier sospecha de RAM a su Centro Autonómico o Regional de Farmacovigilancia mediante las ‘tarjetas amarillas’. Consulte en este directorio su Centro Autonómico de Farmacovigilancia correspondiente.
MÉTODO electrónico: desde el 15 de enero de 2013 se puede notificar a través del sitio web https://www.notificaRAM.es/, y el sistema electrónico hace llegar a su centro correspondiente la notificación de sospecha de RAM. Sirve para profesionales sanitarios y para ciudadanos, en formularios diferentes. La nueva legislación europea de farmacovigilancia establece esta posibilidad para facilitar la notificación de las sospechas de RAM por la población en general.
¿Dónde conseguir tarjetas amarillas?
Consultando a su Centro correspondiente del SEFV-H. Podrá encontrar el directorio de Centros en las primeras páginas del “Catálogo de Medicamentos” y en las páginas de Internet http://www.portalfarma.com y http://www.aemps.gob.es/vigilancia/medicamentosUsoHumano/docs/dir_serfv.pdf.
¿Dónde consultar las fichas técnicas y prospectos de los medicamentos?
En la página web de la AEMPS http://www.aemps.gob.es, seleccionando ”CIMA: Centro de Información on-line de Medicamentos de la AEMPS, Humanos”, se pueden consultar por nombre comercial o por sus principios activos. También están disponibles en la base de datos BOT PLUS.
NOTA: la mención de marcas comerciales en el texto solo tiene fines de identificación, y en absoluto se les debe asignar directamente lo descrito en el texto.





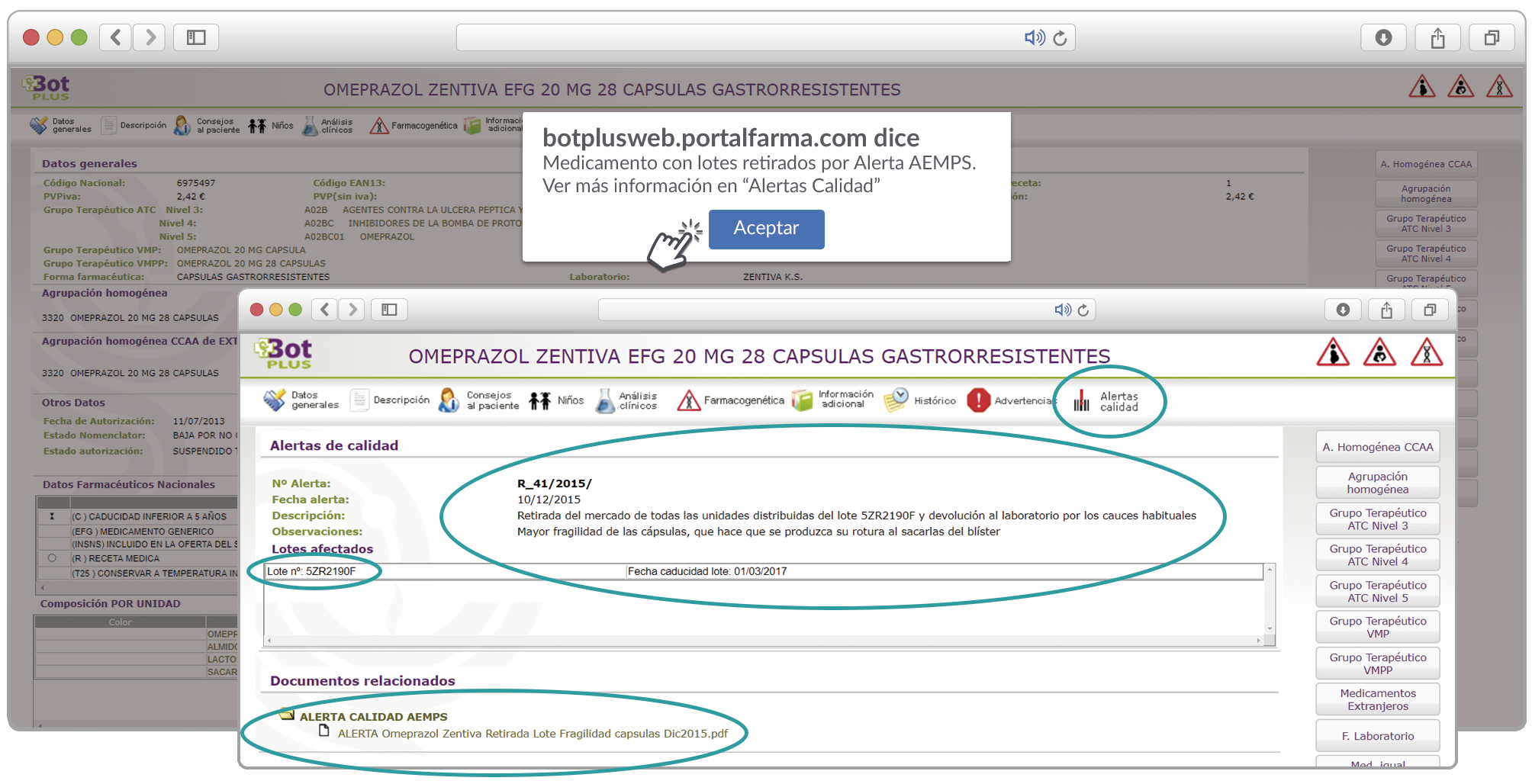
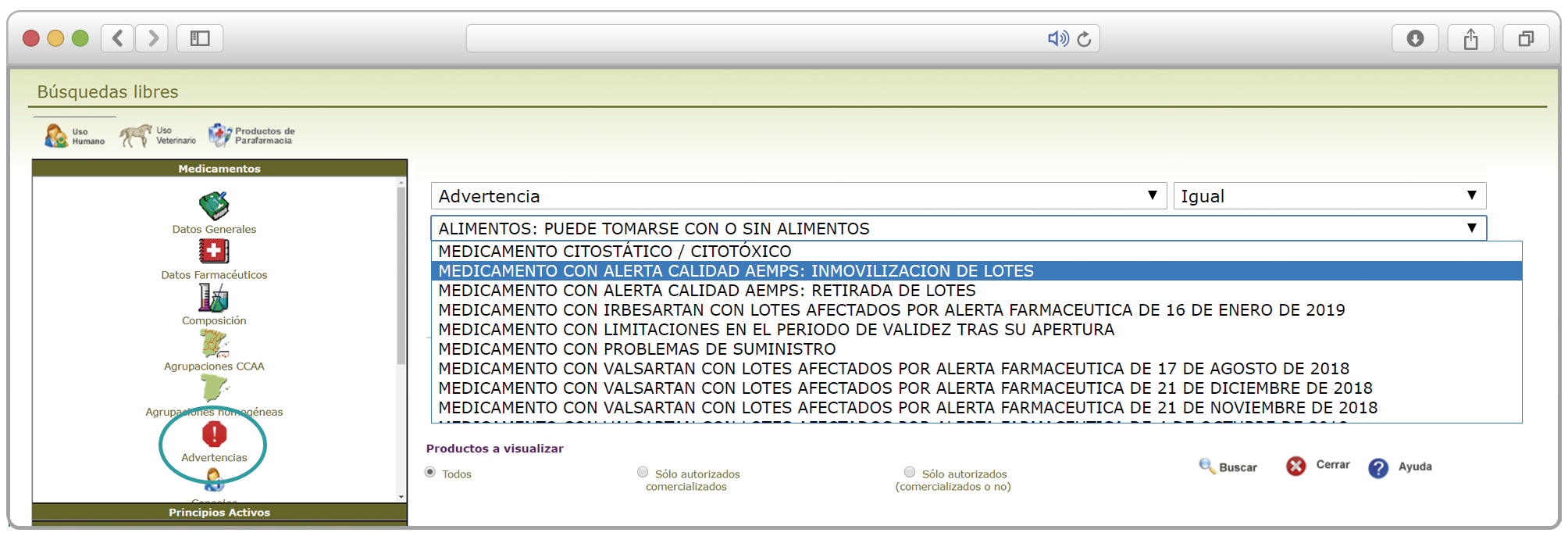
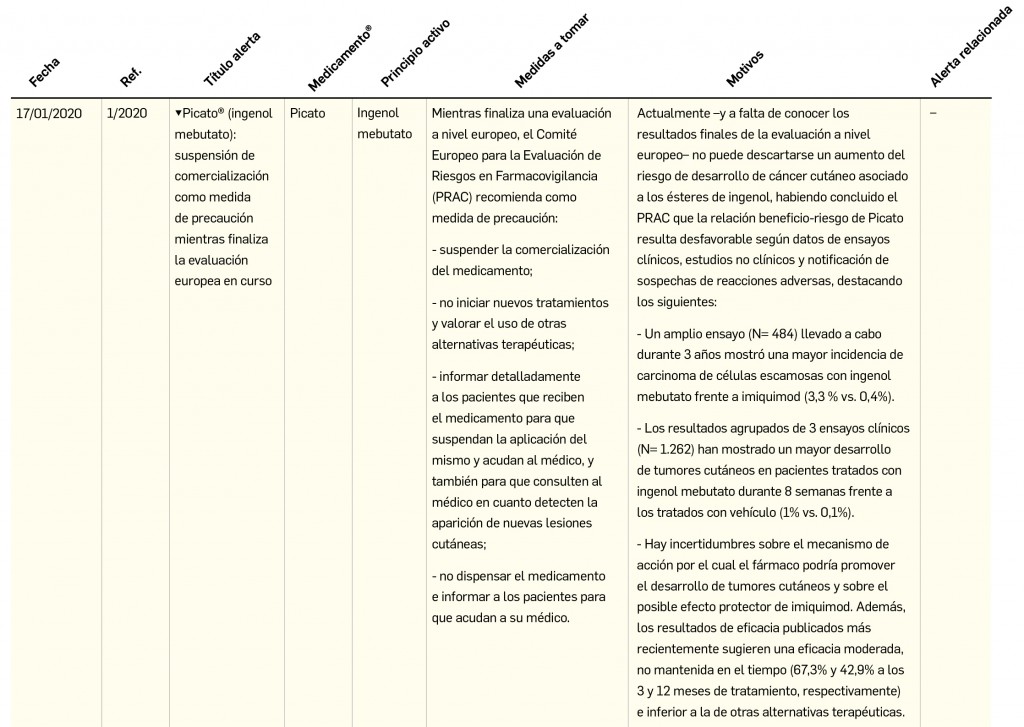
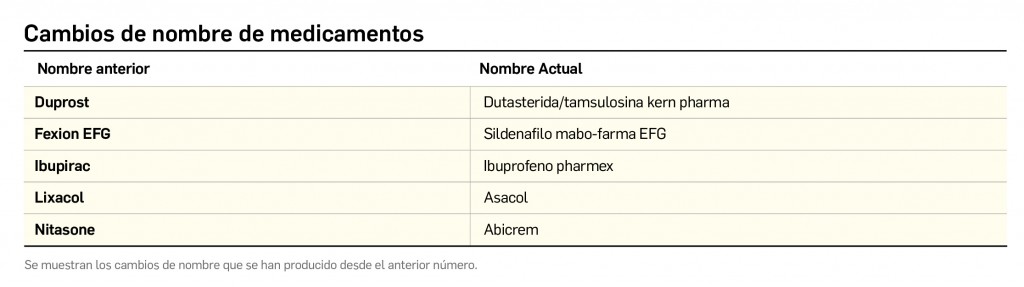
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}