> Ganador, 8ª Edición Premios FORO de Atención Farmacéutica en Farmacia Comunitaria.
Presentación inicial del caso
Se trata de una mujer de 72 años diagnosticada de diabetes mellitus tipo 2 (DM2) desde hace 10 años, que además padece hipertensión arterial (HTA) y poliartritis. Presenta malestar general, edema en las piernas y comenta que ha sufrido durante la semana anterior episodios de mareos con calambres, temblores, sudoración, deposiciones diarreicas frecuentes y pérdida de apetito. Ha incrementado la dosis de ibuprofeno 600 mg a 4 sobres al día debido al dolor por la artritis y lleva una vida sedentaria. No fuma ni bebe alcohol.
El tratamiento farmacológico que tiene pautado es: gliclazida 30 mg (3 comprimidos al día), metformina 850 mg (2 comprimidos al día), enalapril/hidroclorotiazida 20/12,5 mg (1 comprimido al día) e ibuprofeno, si precisa.
Creemos que la paciente puede sufrir un cuadro de hipoglucemia por descompensación de su DM2, ya que no consta autocontrol por la paciente (BMtest) y la última analítica es de hace tiempo; por ello, le ofrecemos un análisis de la glucemia preprandial y otro de hemoglobina glicosilada (HbA1c), además de un control de la presión arterial (PA), colesterol, peso y cálculo del índice de masa corporal (IMC).
Primera vista: estado de situación inicial
Tras una serie de preguntas, observamos un total desconocimiento del tratamiento farmacológico por parte de la paciente. Nos explica que, últimamente, sus valores de PA han estado siempre altos y que cada vez le duelen más las articulaciones. Reitera que durante el año 2017 tuvo muchos problemas de hiperglucemia, razón por la cual el médico le cambió el tratamiento; desde entonces, ha estado bien. La paciente no estaba instruida en cuáles eran los síntomas de una hipoglucemia. Precisamente, pensaba que, por su historial, estaba teniendo una crisis hiperglucémica y la estaba afrontando mediante una reducción excesiva del consumo de azúcares.
Tras la primera entrevista farmacéutica y la revisión de la bolsa de medicamentos, junto con la receta electrónica, observamos que tiene prescrito un tratamiento hipoglucemiante muy intensivo desde el año 2017.
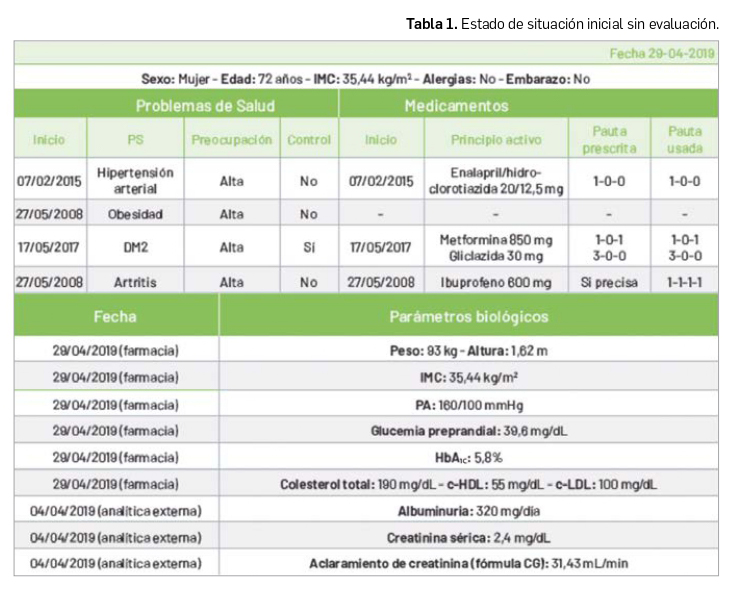
Además de las pruebas realizadas en la farmacia, le pedimos su última analítica (Tabla 1), en la que podemos observar que tiene valores que reflejan una mala función renal. En principio parece que estamos ante una paciente con un mal control de su enfermedad y es, en este momento, cuando entendemos que estamos ante una paciente con un mal seguimiento.
Estudio del caso
El valor de la HbA1c es 5,8% y, por tanto, se puede considerar que ha tenido un buen control de la glucemia en el último trimestre. Sin embargo, el valor de la glucemia preprandial es de 39,6 mg/dl, indicativo de hipoglucemia. Recordamos que la paciente acudió el primer día a la farmacia con los síntomas representativos de ésta: debilidad, cansancio, temblores, sudores, mareos, etc. Podemos pensar que dicha situación viene dada por una combinación de fármacos hipoglucemiantes.
La paciente presenta insuficiencia renal, tal y como indican los valores de la analítica (albuminuria: 320 mg/día; creatinina sérica: 2,4 mg/dl; aclaramiento de creatinina: 31,43 ml/min). Es un factor a tener en cuenta a la hora de pautar el tratamiento farmacológico y razón por la cual se podría estar potenciando el efecto hipoglucemiante de la metformina y, sobre todo, de la gliclazida. Las interacciones entre IECA-AINE y entre hidroclorotiazida-AINE también pueden aumentar el riesgo de empeorar la función renal.
Los valores de PA de 160/100 mmHg sugieren un mal control de la HTA. Parece ser que el tratamiento con IECA + hidroclorotiazida no está siendo efectivo. Además, la combinación IECA-AINE puede producir una disminución del efecto antihipertensivo, con riesgo de pérdida de control de la PA durante el tratamiento prolongado con AINE. Esta situación podría estar produciéndose debido al incremento del uso del ibuprofeno para controlar los dolores derivados de la poliartritis.
Los valores de colesterol total (190 mg/dl), c-HDL (55 mg/dl) y c-LDL (100 mg/dl) se encuentran dentro de los márgenes esperados. La paciente padece obesidad desde hace 9 años, lo cual supone un factor de riesgo en las diferentes patologías que presenta. No ha cambiado su estilo de vida para bajar su IMC.
Evaluación: estado de situación final
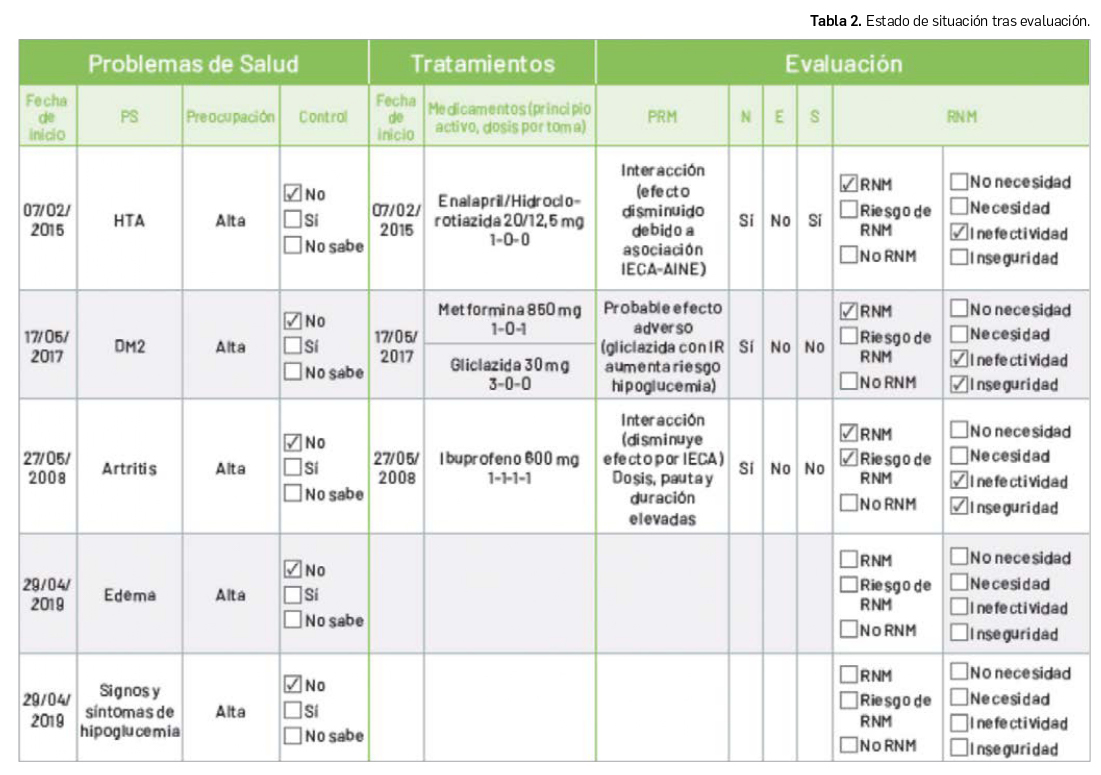
Tras el estudio del caso, y tal y como se ha comentado anteriormente, se detectan diversos Problemas Relacionados con la Medicación (PRM) (Tabla 2). Por un lado, la insuficiencia renal hace que el efecto de la gliclazida se vea potenciado pudiendo dar paso a crisis hipoglucémicas (RNM inseguridad) como la que refiere la paciente. Por otro, la interacción IECA-AINE supone una reducción del efecto antihipertensivo, lo que podría estar relacionado con el mal control de la HTA (RNM inefectividad).
Intervención
Nos ponemos en contacto con el médico, y le explicamos la situación de la paciente. Propusimos un tratamiento alternativo como consecuencia de la evaluación realizada en el Servicio, en base a las siguientes conclusiones:
- Creíamos que lo más oportuno era retirar la gliclazida y reducir la dosis de metformina. También se podría sustituir gliclazida por una gliptina, ya que son una buena alternativa a las sulfonilureas en personas de edad avanzada y con problemas renales.
- Sería conveniente sustituir el IECA por un antagonista de los receptores de la angiotensina II (ARAII) como valsartán, ya que no hay tantos casos descritos de problemas con la insuficiencia renal.
- En relación al diurético, la hidroclorotiazida tampoco está recomendada en insuficiencia renal. Se debería valorar su sustitución.
- Ibuprofeno 600 mg debería ser sustituido por otro analgésico.
A corto plazo nuestra actuación consistió en:
- Recomendar el tratamiento de la hipoglucemia con el consumo de carbohidratos de acción rápida, como tabletas de glucosa o zumos de fruta.
- Derivar al médico para que actualice el tratamiento con los diferentes cambios que habíamos sugerido.
- Ofrecer el servicio diario de la medida de niveles de glucemia preprandial y PA, así como controlar los valores de HTA.
- Preparación de recomendaciones para una dieta individualizada.
Resultados
Después de la consulta con el médico, este modificó la receta electrónica de la paciente siguiendo nuestros consejos y recomendaciones:
- Prescribió metformina 850 mg (1-0-0 en función de las comidas) durante las primeras semanas y luego la sustituyó por Janumet® 50/1000 mg (sitagliptina + metformina).
-
Sustituyó enalapril/hidroclorotiazida 20/12,5 mg por valsartán 80 mg.
- Sustituyó ibuprofeno 600 mg cada 6 horas por paracetamol 650 mg.
Posteriormente, la paciente acude semanalmente a la farmacia para llevar un seguimiento de sus niveles de glucemia, así como de HTA, colesterol y peso. Gracias a la dieta y al cambio de medicación se está consiguiendo reestablecer los valores de estos parámetros dentro de los límites establecidos.
Comentarios
Este caso revela la importancia del farmacéutico comunitario como profesional de la salud. Teniendo en cuenta el plan de medicación, la sintomatología y la última analítica de la paciente, hemos podido observar cómo una disfunción puede potenciar el efecto de los antidiabéticos generando crisis hipoglucémicas. En definitiva, se demuestra cómo el Servicio de Seguimiento Farmacoterapéutico permite detectar problemas y riesgos con la medicación de los pacientes, además de aportar ventajas referentes a la seguridad farmacológica.
Casos ganadores y finalista 8ª edición Premios FORO AF-FC
El objetivo de la edición anual de Premios FORO de Atención Farmacéutica en Farmacia Comunitaria (Foro AF-FC) es reconocer el compromiso asistencial de los farmacéuticos que ofrecen a la población Servicios Profesionales Asistenciales de acuerdo a los procedimientos consensuados1, contando con la colaboración de laboratorios Cinfa. La convocatoria de la 8ª edición se realizó entre los meses de marzo a julio de 2019, periodo tras el cual el jurado, compuesto por seis representantes de las instituciones que forman Foro AF-FC, evaluó los 30 casos recibidos de acuerdo a los modelos editados en las webs de todas las instituciones para la descarga, cumplimentación y remisión on line, por parte de farmacéuticos voluntariamente interesados.
Se mantuvo un modelo abierto para aquellos casos que no se correspondieran con los Servicios descritos. Como novedad, en esta edición se posibilitó una categoría específica para casos de alumnos y tutores en prácticas tuteladas o máster. Los casos recibidos se clasificaron en: 8 casos del Servicio de Dispensación, 1 del Servicio de Indicación Farmacéutica, 3 del Servicio de SFT, 15 de modelo abierto y 4 de alumnos. El jurado valoró en cada caso once apartados relacionados con la adecuación de la terminología y metodología de Foro AF-FC, la existencia de práctica colaborativa, la utilización de herramientas informáticas, el interés clínico y el profesional del caso en la Farmacia Comunitaria, entre otros.
Los casos ganadores y finalistas de la citada edición se irán presentando periódicamente en esta sección de PAM. No obstante, puede acceder a la publicación íntegra de todos los casos a través del siguiente enlace al espacio de los Premios de FORO AF-FC:
Foro de Atención Farmacéutica en Farmacia Comunitaria (Foro AF-FC) es un grupo de trabajo y debate constructivo creado en 2009. Actualmente está constituido por farmacéuticos representantes de las siguientes instituciones:
- Consejo General de Colegios Oficiales de Farmacéuticos (CGCOF).
- Conferencia Nacional de Decanos.
- Fundación Pharmaceutical Care España.
- Grupo de Investigación en Atención Farmacéutica de la Universidad de Granada.
- Sociedad Española de Farmacia Familiar y Comunitaria (SEFAC).
La principal función de Foro AF-FC es contribuir a la implantación de los Servicios Profesionales Farmacéuticos
Asistenciales (SPFA) en la farmacia comunitaria, para lo cual:
- Mantiene el consenso adoptado y la homogeneidad de los procedimientos de Foro AF 2008 en los proyectos, conjunta o individualmente; de esta manera, se consigue un objetivo común transmitiendo el mismo mensaje con una terminología consensuada.
- Apoya la máxima difusión de los SPFA para alcanzar su implantación en la Farmacia Comunitaria.
- Incrementa la colaboración entre las organizaciones del Grupo.
- Constituye un agente de referencia en SPFA coordinado por el Consejo General.
Principales actividades:
- Elaboración de documentos consensuados; definición y procedimientación de Servicios Profesionales Farmacéuticos
- Asistenciales.
- Definición y clasificación de SPFA abordando tanto los relacionados con el medicamento (AF) como con los relacionados con la Salud Comunitaria.
- Edición de la actualización de la Guía practica de SPFA
- Edición anual de Premios a los mejores casos de farmacéuticos implicados en SPFA y participación en eventos/congresos para su divulgacion.
Más información en: http://www.portalfarma.com/inicio/serviciosprofesionales/forofarmaciacomunitaria/Paginas/default.aspx






{kind=link}
{kind=link}