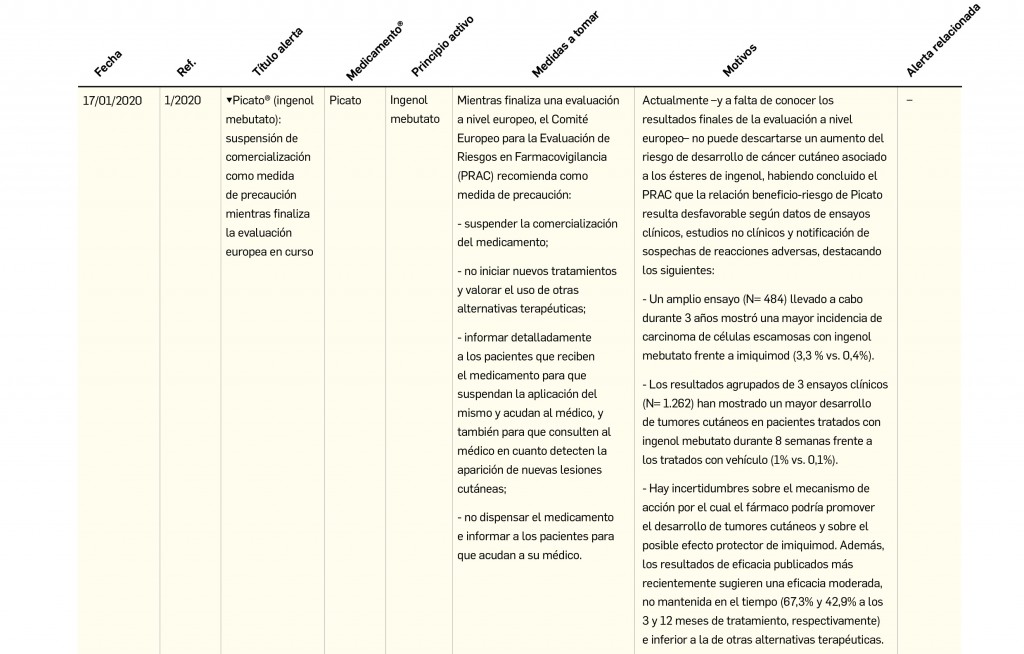
Resumen de las notas sobre seguridad y farmacovigilancia publicadas por la AEMPS desde principios del año 2020. Para información más ampliada y acceso al documento de la AEMPS, puede consultar BOT PLUS.
Número 434, Junio 2020
Resumen de las notas sobre seguridad y farmacovigilancia publicadas por la AEMPS desde principios del año 2020. Para información más ampliada y acceso al documento de la AEMPS, puede consultar BOT PLUS.
Además de la información que se incluye en los listados mensuales publicados en PAM, en BOT PLUS se incluye un apartado de Histórico, en las fichas de medicamentos, en el que se presenta información referente a cambios que haya sufrido anteriormente el medicamento o producto, entre otros, los cambios de nombre y los cambios de laboratorio. Esta información también está disponible para productos sanitarios financiados o dietoterápicos.
Se añade la posibilidad de visualización de las situaciones anteriores (o incluso futuras) relacionadas con un cambio de nombre.
Con automatismos que nos permiten localizar un medicamento que haya cambiado de nombre, independientemente de cuál usemos.
Además de la información existente en Histórico, se permite la explotación de la información incluida en BOT PLUS en este apartado, mediante la integración de la información almacenada en Histórico en el apartado de Listados de BOT PLUS, que permite realizar consultas entre rangos de fechas y por un concepto en concreto de entre los almacenados en el apartado de Histórico. Entre ellos se incluyen, precisamente, los conceptos “Cambio del nombre del medicamento” y “Cambio del laboratorio comercializador”.
_______________________________________________________________________________________________
Valoración de la innovación terapéutica en PAM
Es importante indicar que se valora el grado de innovación. Todos los medicamentos, sean innovadores o no, tienen utilidad terapéutica, en tanto que su autorización por las autoridades sanitarias implica que han demostrado rigurosamente su eficacia, su seguridad, su calidad y las condiciones de uso (incluyendo la información contenida en la ficha técnica – sumario de características – y en el prospecto del medicamento). Por tanto, la valoración que se hace se refiere a la incorporación, en el grado que se determine, de algún elemento innovador con respecto a otros medicamentos autorizados previamente para iguales o similares indicaciones terapéuticas o, en su caso, cubriendo la ausencia de éstas.
Asimismo, debe considerarse que ésta es una evaluación que se practica coincidiendo con la comercialización inicial del medicamento. Se trata, por consiguiente, de una valoración provisional de la innovación realizada en función de la evidencia clínica disponible hasta el momento, lo que no prejuzga, en ningún caso, la disponibilidad posterior de nuevas evidencias científicas (de eficacia o de seguridad) en la indicación autorizada o el potencial desarrollo y autorización, en su caso, de nuevas indicaciones terapéuticas o la imposición de restricciones de uso en las anteriores.
Se consideran tres posibles niveles, adjudicados en función de la relevancia de la(s) innovación(es) presentes en el nuevo medicamento, siempre en relación al arsenal terapéutico disponible clínicamente en España en el momento de la comercialización:
Se distinguen dos niveles de evidencia científica para los aspectos innovadores de los nuevos medicamentos:
El rigor de los datos contrastados mediante ensayos clínicos controlados (evidencia clínica) es determinante en la valoración de la innovación, mientras que las potencialidades solo pueden ser valoradas accesoriamente, como aspectos complementarios de esta valoración. En ningún caso, un medicamento es valorado con un nivel de innovación importante en función de sus ventajas potenciales, si no aporta otras ventajas demostradas clínicamente. Se analizan cinco aspectos de la innovación: clínica, molecular, toxicológica, físico-química y económico-tecnológica. Como ya se ha indicado, la fundamental y determinante es la novedad clínica.
Inotersén es un oligonucleótido antisentido diseñado para su unión selectiva a la región no traducida en 3’ del ARNm de la transtiretina (TTR) humana, tanto de tipo mutante como normal o salvaje, y provoca su degradación a través de la escisión mediada por la RNAsa H1. Por su parte, patisirán es el primer ARN de interferencia autorizado en Europa: un ARN bicatenario pequeño formulado en nanopartículas lipídicas que se une específicamente a una secuencia conservada en la región 3’-UTR del ARNm de la TTR, también tanto en su forma mutante como salvaje, y a través de la interferencia de ARN y con mediación de la endonucleasa argonauta-2, produce la degradación catalítica de dicho ARNm. En consecuencia, ambos fármacos inhiben la síntesis hepática y la secreción a sangre de la proteína TTR, reduciendo de forma sustancial sus niveles séricos circulantes, lo que se traduce en una mayor estabilización o aclaramiento de los depósitos de TTR amiloidótica y de las manifestaciones de la polineuropatía. Designados por la EMA como medicamentos huérfanos, ambos han sido autorizados para el tratamiento de la polineuropatía en estadios 1 o 2 en pacientes adultos con amiloidosis familiar (o hereditaria) por transtiretina (ATTRh). Inotersén se administra por vía subcutánea una vez por semana y patisirán por vía intravenosa una vez cada 3 semanas.
La eficacia y seguridad clínicas de inotersén han sido contrastadas en un ensayo aleatorizado de fase 2/3 (N= 173), doble ciego y controlado por placebo. El tratamiento durante 15 meses demostró una eficacia en la estabilización de los síntomas neurológicos periféricos significativamente superior a placebo (diferencia de 15 puntos en la escala mNIS+7) y en la calidad de vida de los pacientes (diferencia de 9 puntos en el cuestionario Norfolk QoL-DN). De forma similar, la superioridad de patisirán frente a placebo para frenar la lesión nerviosa progresiva fue demostrada en un ensayo pivotal de fase 3 (N= 225), también doble ciego y aleatorizado. Tras 18 meses, el fármaco redujo una media de 6 puntos la puntuación de mNIS+7 (diferencia de 34 puntos respecto a placebo), indicando una ligera mejoría de los pacientes, significativa desde el 9º mes de tratamiento; también indujo una modesta mejora de su calidad de vida (diferencia de 21 puntos en el cuestionario Norfolk QoL-DN a favor de patisirán). Adicionalmente, las variables secundarias respaldaron el beneficio aportado por los dos fármacos sobre la fuerza motora, el estado nutricional o la velocidad de la marcha; una eficacia que se mostraba consistente entre subgrupos de pacientes e independiente de factores como el tipo de mutación, gravedad del paciente, tratamiento previo o presencia de cardiopatía.
El perfil toxicológico a corto-medio plazo de ambos fármacos, más benigno para patisirán, es clínicamente manejable, siendo la mayoría de eventos adversos leves-moderados y autolimitados. Las reacciones adversas más frecuentes con inotersén son reacciones en el lugar de inyección (51%) –sobre todo, eritema, dolor y prurito–, náuseas (31%), anemia (28%) o cefalea (23%), si bien sobresale, por su gravedad, el riesgo de trombocitopenia (13%, grave en el 7%) y de glomerulonefritis (3%), así como una inmunogenicidad no desdeñable. Los eventos adversos más comunes con el mejor tolerado patisirán son: edema periférico (30%) y reacciones relacionadas con la perfusión (19%), como dolor de espalda, rubefacción, dolor abdominal o náuseas. Los estudios de extensión ahora en marcha aportarán más datos a largo plazo.
En definitiva, son dos nuevos tratamientos etiológicos que inauguran una vía terapéutica en el tratamiento de la ATTRh y aportan un beneficio clínicamente relevante en el manejo de la polineuropatía: aunque se exceptúan los pacientes más graves (estadio 3), amplían las posibilidades terapéuticas de tafamidis (pacientes en estadio 1 exclusivamente) y serán opciones de tratamiento útiles, especialmente si se considera que hasta dos tercios de los pacientes no son susceptibles de trasplante hepático. No se dispone aún de comparaciones directas o indirectas entre las distintas opciones de tratamiento ni se puede concluir acerca de la superioridad de una sobre otra, pero los resultados divulgados podrían sugerir que inotersén y patisirán son más eficaces que tafamidis. Patisirán parece aportar un mayor beneficio clínico y una mayor innovación terapéutica.
La amiloidosis transtiretina (ATTR) es un trastorno sistémico, progresivamente degenerativo e irreversible, caracterizado por la deposición extracelular de fibrillas amiloides compuestas de monómeros anómalos de transtiretina (TTR).
La TTR, también conocida como prealbúmina, es una proteína que se sintetiza predominantemente en el hígado (> 95%, pero también una parte en el plexo coroideo y la retina) y en su forma fisiológica se dispone en forma de agregados solubles de cuatro moléculas (tetrámeros); desempeña un papel vital en el plasma humano y líquido cefalorraquídeo, actuando como un portador secundario de la tiroxina (T4), el único portador de la vitamina A mediante la proteína de unión a retinol (RBP) en plasma y el principal transportista de la T4 en el LCR. La TTR es una de las aproximadamente 30 proteínas amiloidogénicas humanas relacionadas con enfermedades causadas por fibrillas de amiloide.
Se considera que la disociación del tetrámero de transtiretina en monómeros es el paso determinante de la patogenia de la amiloidosis trantiretina, cuya manifestación clínica más común es la polineuropatía amiloidótica por TTR, también conocida como polineuropatía amiloidótica familiar por TTR (PAF-TTR)1. Los monómeros inadecuadamente plegados son susceptibles de experimentar un proceso de desnaturalización, facilitando posteriormente la formación de estructuras de tipo amiloide, mediante el ensamblaje de dichos monómeros, lo cual da lugar a pequeños agregados u oligómeros solubles, responsables directos de la formación de filamentos y fibrillas de amiloide. Estas estructuras fibrilares, muy estables, se depositan en diversos órganos y tejidos (en particular, en sistema nervioso periférico, tracto gastrointestinal, riñón y corazón), causando muerte celular y disfunción progresiva. La presentación fenotípica de la enfermedad depende fundamentalmente del patrón de órganos afectados: la mayoría de pacientes muestra cierto grado de signos y síntomas de patología en distintos sistemas; se trata, por tanto, de una misma enfermedad con distintos subtipos, siendo las más frecuentes la neuropatía y la cardiomioptaía.
La amiloidosis transtiretina es la forma más común de amiloidosis familiar y es causada por mutaciones en el gen codificante para la TTR, que resultan en la desestabilización de la proteína. Esta forma de amiloidosis también incluye a la amiloidosis sistémica senil, una forma de amiloidosis relacionada con la edad y de carácter adquirido, que afecta principalmente a los hombres después de la edad de 60 años (Ando et al., 2013).
La amiloidosis hereditaria (o familiar) por TTR tiene una herencia autosómica dominante con un grado de penetración variable. Los portadores de la mutación (la mayoría hetereocigotos) tienen una variante anómala de la proteína circulante desde la vida fetal, pero sin que se produzca la deposición de amiloide ni se manifieste la enfermedad sintomática generalmente hasta la edad adulta, cuyo desarrollo posiblemente está relacionado con factores ligados con la bioquímica normal del propio envejecimiento. Algunos datos sugieren que las mujeres afectadas tienen una mayor capacidad para transmitir la enfermedad a su descendencia que los hombres afectados. En cualquier caso, ciertos portadores del gen pueden vivir hasta una edad avanzada sin síntomas de la enfermedad, pero al mismo tiempo algunos niños pueden presentar signos clínicos.
En 1942 se registró en Portugal un amplio grupo de pacientes con la mutación más característica de la enfermedad: la Val30Met. Durante las siguientes dos décadas fueron descubiertos otros importantes colectivos en Japón y Suecia. Hasta la fecha, se han descrito aproximadamente 120 diferentes mutaciones simples, dobles o por deleción del gen codificante para TTR (18q12.1), la mayoría amiloidogénicas y, de hecho, menos de diez se consideran no patógenas. Algunas mutaciones inducen miocardiopatía como rasgo patológico predominante (Val122Ile, Ile68Leu, Thr60Ala, Leu111Met), mientras que otras están asociadas principalmente con la neuropatía, particularmente la Val30Met, pero ambas manifestaciones pueden estar presentes en diferentes proporciones en los pacientes. Otros signos menos comunes incluyen opacidad vítrea, enfermedad renal y afectación meníngea. El cuadro clínico de un paciente puede deteriorarse con el tiempo a medida que el amiloide se sigue depositando en los diversos tejidos e incluso puede observarse variabilidad dentro de la misma familia.
La prevalencia a nivel mundial de amiloidosis hereditaria por TTR con polineuropatía se estima en aproximadamente 10.000 pacientes (Coelho et al., 2008), mientras que en la Unión Europea se calcula que la población actual de pacientes es de 2.700 a 3.500 casos (prevalencia de 0,52 casos por 100.000 habitantes). Si se habla de amiloidosis hereditaria por TTR en general, algunos estudios apuntan a que la prevalencia estimada en Europa es de 0,1 pacientes por cada 10.000 habitantes, lo que totalizaría entre 5.000 y 6.000 casos, la mayoría de ellos en Portugal, Francia, Italia y Reino Unido. En todo caso, parece evidente que estamos ante una enfermedad rara con predominancia en varones sobre mujeres (ratio 3:1).
En Europa, se cree que las mutaciones del gen TTR acontecen en menos de 1 de cada 100.000 personas (incidencia estimada de 0,03 casos por cada 100.000 habitantes y por año), pero en áreas endémicas del norte de Suecia la prevalencia de la mutación Val30Met llega a ser del 4%, aunque las manifestaciones clínicas son relativamente infrecuentes (11% en portadores de 50 años); en cambio, en Portugal, la presentación clínica es alta (80% en los portadores de 50 años). Aunque también es endémica en algunas zonas de Japón, la prevalencia general de la enfermedad se estima que es menor que en Europa, siendo aproximadamente de un caso por cada millón personas (Coelho et al., 2016). En contraste, la incidencia de la amiloidosis hereditaria por TTR en los Estados Unidos se estima en 1:100.000.
Como se ha sugerido, la mutación Val30Met es la más comúnmente detectada en la amiloidosis hereditaria por TTR en el mundo (representa más de la mitad de todos los casos) y la única que se encuentra en grandes grupos de pacientes, con especial relevancia en Portugal, Suecia, Japón, Brasil y, dentro de España, en Islas Baleares2. El mayor grupo de personas con Val30Met se puede encontrar en el norte de Portugal, donde se estima que la incidencia es de 1:538. Las mutaciones Leu111Met y Val122Ile (relacionadas con cardiomiopatía) se encuentran principalmente en las poblaciones danesas y afroamericanas, respectivamente.
La edad de inicio de los síntomas relacionados con la enfermedad varía ampliamente entre las diferentes poblaciones, desde la segunda a la novena década de la vida, siendo mayoritaria la aparición en la séptima década. Las poblaciones portuguesas y japonesas de pacientes con amiloidosis hereditaria por TTR tradicionalmente tienen un debut clínico temprano (33 años de edad, de promedio), mientras que los pacientes suecos se caracterizan por una media de edad de inicio más tardía (56 años); incluso en las poblaciones de inicio temprano, algunos subgrupos de pacientes tienen un debut clínico más tardío. En general, la velocidad de progresión de la enfermedad suele ser más rápida en aquellos casos de aparición más temprana, que tendrán una menor supervivencia. La duración media del periodo comprendido entre la aparición de la enfermedad hasta la muerte puede variar dependiendo de la zona endémica, el genotipo, los síntomas (la presencia de una cardiomiopatía significativa se asocia con un peor pronóstico) y otros factores. Algunos autores hablan de que puede estar en torno a los 10,8 años, si bien otros trabajos apuntan a una mediana de supervivencia de 4,7 años o 5,5 años (Swiecky et al., 2015) en pacientes con cardiomiopatía.
Se trata de una enfermedad con múltiples y muy diversos perfiles clínicos, pudiendo presentarse neuropatía periférica (sensitiva y motora), neuropatía autonómica, alteraciones gastrointestinales, cardiomiopatía, nefropatía u opacidad ocular. Una mayoría de los casos se clasifican como polineuropáticos. Los síntomas de la neuropatía sensorial y motora de las extremidades pueden presentarse en un plazo corto o incluso al mismo tiempo, mientras que la neuropatía autonómica puede ser relativamente leve. No obstante, hay que subrayar que el cuadro clínico de los portadores de Val30Met difiere considerablemente entre los pacientes procedentes de focos endémicos y aquellos con orígenes no endémicos. En los primeros, la enfermedad suele debutar antes de los 40 años de edad, con alteraciones sensorio-motoras progresivas y neuropatía autonómica, causando finalmente caquexia y la muerte en torno a 10 años después de su inicio. Por su parte, los pacientes de origen no endémico tienen generalmente inicio a una edad tardía (> 60 años), con predominio del sexo masculino y una presentación de la enfermedad aparentemente esporádica.
La amiloidosis hereditaria (o familiar) por TTR induce una neuropatía periférica de carácter progresivo. En una primera etapa, dado que el amiloide típicamente afecta primero pequeñas fibras nerviosas, se reduce la sensibilidad dolorosa y la térmica, particularmente en los pies. Por el contrario, la sensibilidad a la luz y la propiocepción pueden estar relativamente preservadas; la fuerza muscular y los reflejos tendinosos son también normales. Conforme pasan algunos meses, la pérdida sensorial se extiende por encima del tobillo en ambos lados, y más adelante, el déficit neurológico progresa implacablemente, con la extensión de la pérdida de la sensibilidad hacia las zonas proximales de las extremidades inferiores. El déficit motor por atrofia muscular aparece en los miembros inferiores distales, haciendo que el caminar se vuelva cada vez más difícil, debido a la pérdida del equilibrio, dando a la marcha una forma característica (marcha equina). El dolor neuropático es a menudo de tipo pungente (como una quemadura), siendo peor en la noche y se asocia con alodinia (sensación dolorosa exagerada en respuesta pequeños estímulos). Con el transcurrir del tiempo, el déficit sensorial se extiende a los muslos y miembros superiores, en particular antebrazos y dedos. Los déficits motores también siguen una progresión dependiente de la duración, y caminar sin ayuda se vuelve cada vez más difícil: los pacientes normalmente terminan en silla de ruedas. La pérdida de la sensación de dolor con la preservación de la fuerza conduce a traumas no dolorosos y al desarrollo de úlceras plantares y osteoartropatía en los pies (articulaciones de Charcot). El síndrome del túnel carpiano es una manifestación temprana, aunque no específica.
La alteración del sistema nervioso autónomo –neuropatía autonómica– que se produce en muchos pacientes incluye anhidrosis, impotencia sexual, trastornos de la motilidad gastrointestinal (el más común es la diarrea, que se alterna con estreñimiento, náuseas y vómitos), hipotensión ortostática y vejiga neurogénica. La enfermedad cardíaca3 se produce en aproximadamente el 50% de los pacientes (hasta en el 80% en algunos grupos), y la anemia debida a los bajos niveles de eritropoyetina también se observa con relativa frecuencia. La afectación ocular, manifestada como opacidad del vítreo, ojo seco, glaucoma y trastornos de la pupila, es común, mientras que el deterioro renal es infrecuente. Otros síntomas incluyen: ronquera, disminución de la temperatura de la piel, disestesia, debilidad muscular y atrofia, anestesia disociada, pérdida de peso, arritmias, edemas y sensación de quemazón.
Por otro lado, existe una forma leptomeníngea relativamente rara –pero grave– de la enfermedad, inducida por varias mutaciones puntuales en el gen TTR, incluyendo la Val30Met. La angiopatía amiloide cerebral y la amiloidosis ocular son las características clínicas comunes de este tipo de amiloidosis, incluyendo entre sus manifestaciones neurológicas más típicas el infarto cerebral y hemorragia, hidrocefalia, ataxia, parálisis espástica, convulsiones y demencia. Finalmente, asociados a la neuropatía autonómica, se describen desde las fases iniciales los síntomas gastrointestinales: náuseas, saciedad precoz, vómitos recurrentes, diarrea acuosa, estreñimiento intenso y/o diarrea y estreñimiento alternados; como resultado de ello, es fácil observar una progresiva pérdida de peso a lo largo del curso de la enfermedad.
En vista de lo anterior, se entiende que la severidad de la patología provoca un elevado impacto sobre la calidad de vida de los pacientes y de sus cuidadores; estos últimos presentan niveles moderados-altos de fatiga e invierten una parte importante de su tiempo en el cuidado de los pacientes. Así, la amiloidosis hereditaria por TTR se asocia con una reducción sustancial de la tasa de empleo y productividad, así como con un alto impacto socio-psicológico en pacientes y cuidadores.
Las opciones terapéuticas actuales para los pacientes con amiloidosis hereditaria (o familiar) por TTR son escasas y claramente limitadas. Para aquellos pacientes con diagnóstico confirmado por clínica, pruebas genéticas y biopsia (confirmación de los depósitos de amiloide en la grasa abdominal, glándulas salivares o en el recto), y enfermedad leve o moderada –aquellos que inician su enfermedad antes de los 50 años y tienen historia corta de manifestaciones–, el trasplante de hígado es el tratamiento estándar actual. El trasplante debe eliminar casi por completo la producción de la variante anómala de la proteína (que será el efecto farmacológico crucial en el beneficio clínico) y detener así la progresión de la enfermedad, dado que es el mayor órgano productor de TTR; no obstante, la TTR normal se seguirá produciendo por el órgano implantado y puede depositarse en los depósitos de amiloide pre-existentes en los tejidos, con progresión de la enfermedad.
Por los requerimientos previos, casi dos tercios de los pacientes no son candidatos a trasplante. Entre quienes se realiza, el trasplante consigue estabilizar la progresión de la neuropatía sensorial y motora en aproximadamente el 60% de los pacientes, principalmente en los receptores de trasplante con la mutación Val30Met; un 20% mejoran, aunque la reversibilidad es extremadamente rara. El trasplante se relaciona con una mejora de la supervivencia de los pacientes, siendo la supervivencia a los 10 años del 71%, aunque la mortalidad durante el primer año postrasplante es del 10%. Sin embargo, esta opción no impide eficazmente la cardiomiopatía en muchos casos, ni tampoco mejora los síntomas oculares o del sistema nervioso central, requiere la administración crónica de inmunosupresores, y no se recomienda para los pacientes con la fase final de la amiloidosis por TTR o con la forma leptomeníngea. Además, incluso entre un 15-20% de los pacientes empeoran tras el trasplante.
Para todos los pacientes, los tratamientos sintomáticos son esenciales para ayudar a controlar los síntomas asociados a las neuropatías sensitivo-motoras y autonómicas y las complicaciones viscerales pues, tal como se ha dicho, la muerte sobreviene al cabo de unos pocos años después de la aparición de los primeros síntomas y puede ocurrir repentinamente o derivada de infecciones, malnutrición o caquexia, aunque posiblemente también de fallo renal o patología cardiaca (Coelho et al., 2008). En general, el tratamiento de soporte estándar requiere un abordaje multidisciplinar que involucra a neurología, gastroenterología y cardiología, debiendo instaurarse fundamentalmente tratamientos paliativos para el dolor, náuseas, vómitos y diarrea, si bien su eficacia será limitada por la progresión de la enfermedad.
La elucidación de la estructura del tetrámero y los mecanismos que contribuyen al plegamiento de la transtiretina y a la formación de las fibrillas proteicas ha permitido identificar el proceso de estabilización del tetrámero original como un elemento limitante de la velocidad de formación de las fibrillas amiloides. En este sentido, se desarrollaron cálculos mecano-cuánticos que permitieron definir series de posibles candidatos para estabilizar dicha estructura, que pudieran ser utilizados durante las etapas tempranas de la enfermedad para evitar la necesidad de trasplante de hígado o, al menos, retrasarla.
Por ejemplo, tafamidis (Vyndaqel®) es un agente modificador de la enfermedad que estabiliza cinéticamente a la transtiretina, uniéndose al sitio de unión de la tiroxina y limitando la disociación del tetrámero en monómeros, un paso crítico en la generación de fibrillas: al ser capaz de prevenir la disociación de los tetrámeros naturales o mutados de transtiretina en sus monómeros, impide la formación de las fibrillas amiloides causantes de la amiloidosis. Autorizado en 2011 por la EMA en circunstancias especiales, está indicado en el tratamiento de la amiloidosis por transtiretina en pacientes adultos con polineuropatía sintomática en estadio 1 (progresión leve, los pacientes no requieren ayuda para caminar) para retrasar la alteración neurológica periférica, ya que en su ensayo pivotal mostró mejoría o estabilización significativa de los síntomas neurológicos en el 45% de pacientes vs. 30% con placebo; asimismo, el tratamiento con tafamidis aporta un beneficio sobre la calidad de vida de los pacientes y su estatus nutricional. No obstante, además de la modestia en esos resultados, tafamidis no parece aportar una mejoría significativa en biomarcadores cardiacos en pacientes con cardiopatía infiltrativa (derivada de la amiloidosis por transtiretina), y el seguimiento a largo plazo de los pacientes que continuaron el tratamiento demuestra que la neuropatía progresa con el tiempo. Su perfil toxicológico no parece ser especialmente preocupante, a pesar de que se aprecia un incremento de las infecciones de tracto urogenital y de diarrea, manejables con tratamientos convencionales (Cuéllar, 2014).
Asimismo, el diflunisal, un antiguo AINE ampliamente conocido, actúa similar a tafamidis: estabiliza in vitro los tetrámeros de TTR impidiendo su desagregación, la liberación de monómeros y la formación de fibrillas de amiloide por monómeros TTR mal plegados. Aunque sin efecto sobre las manifestaciones cardíacas, ha demostrado capacidad para reducir la progresión de la neuropatía en comparación con placebo, tanto en pacientes con estados iniciales como más avanzados, con mutaciones Val30Met y otras diferentes, si bien no ha sido autorizado en ningún país y se hace de él un uso off label. La combinación de doxiciclina y ácido tauroursodeoxicólico también altera la formación de fibrillas de amiloide y puede reducir la deposición de TTR no fibrilar.
En todo caso, la amiloidosis hereditaria (o familiar) por TTR sigue siendo una enfermedad rara frente a la cual se necesitan tratamientos –especialmente para pacientes en estadios avanzados– que, aunque no sean curativos (opción que, en principio, solo podría llegar con la terapia génica), mejoren los resultados clínicos, el pronóstico y la esperanza de vida. Otras líneas de investigación interesantes que ahora dan sus frutos, como comentaremos a continuación, han sido el empleo de moléculas o nucleótidos antisentido y ARN de interferencia diseñados para suprimir la expresión de la proteína TTR y prevenir la formación de amiloides.
Conviene recordar, por último, que en la clasificación y en los ensayos clínicos de opciones terapéuticas frente a esta enfermedad se valoran varios estadios de la polineuropatía en relación con la movilidad:
Adicionalmente, la gravedad de la enfermedad se puede evaluar utilizando la puntuación de discapacidad de polineuropatía (PND por sus siglas en inglés, Polyneuropathy Disability Score), que es una escala de valoración en 5 grados: 0- normal, 1- trastornos sensoriales en las extremidades con capacidad para caminar preservada, 2- dificultad para caminar pero sin necesidad de un apoyo, 3a- requiere un apoyo (palo, bastón) para caminar, 3b- requiere dos apoyos para caminar, y 4- en silla de ruedas o en cama. También es común el empleo del índice de deterioro neurológico (NIS, Neurological Impairment Score); por ejemplo, en pacientes en que la polineuropatía se diagnostica cuando ya tiene al menos en severidad moderada, la progresión es relativamente rápida y, en tratamiento con placebo, suele incrementarse 10-14 puntos de deterioro neurológico por año (lo cual contrasta, por ejemplo, con la polineuropatía diabética, en que la progresión de la puntuación de NIS suele ser menor de 1 punto/año).
Inotersén es un oligonucleótido antisentido que inhibe la producción de transtiretina (TTR) humana mediante su unión selectiva (con una especificidad del 100%) a la región 3´ no traducida (3’-UTR, por sus siglas en inglés) del ARN mensajero (ARNm) de la TTR, lo cual provoca la degradación –a través de la escisión mediada por la RNAsa H1– del ARNm de la TTR tanto de tipo mutante como salvaje o normal. Ejerce así una inhibición concentración-dependiente de la síntesis hepática de la proteína y su secreción a sangre, que se traduce en una reducción sustancial de los niveles circulantes de TTR, tanto de tipo mutado como salvaje, y con ello, en una mayor estabilización o aclaramiento de los depósitos de TTR amiloidótica y de las manifestaciones de la polineuropatía y cardiomiopatía. En base a lo anterior, el medicamento ha sido autorizado para el tratamiento de polineuropatía en estadio 1 o estadio 2 en pacientes adultos con amiloidosis familiar por transtiretina (ATTR).
En ensayos clínicos, se ha probado que en pacientes tratados con inotersén durante 15 meses, los niveles circulantes de TTR se reducían respecto al estado basal en un 68-74% (en comparación con el tratamiento con placebo, que mostraba reducciones de un 8,5% en la concentración sérica media de TTR respecto al estado basal). Sin embargo, no se conoce con seguridad cuál es la reducción mínima en los niveles de TTR para generar un beneficio clínico en los pacientes (EMA, 2018b).
Por su parte, patisirán es un ácido ribonucleico pequeño de interferencia (siRNA, por sus siglas en inglés) bicatenario diseñado para unirse específicamente a una secuencia conservada genéticamente en la región 3’ no traducida del ARNm de la proteína TTR, tanto en su forma mutante como salvaje. Se formula en nanopartículas con 4 excipientes lipídicos que protegen al fármaco de la acción de endo- y exonucleasas en la circulación sistémica y facilitan su liberación en el hígado: el patisirán, una vez en los hepatocitos, actúa a través del proceso natural denominado interferencia de ARN y produce la degradación catalítica del ARNm de la TTR humana (Figura 1), reduciendo sus niveles séricos. El medicamento ha sido autorizado para el tratamiento de la amiloidosis hereditaria por transtiretina (ATTRh) en pacientes adultos con polineuropatía en estadio 1 o 2.
De forma similar a lo comentado para inotersén, patisirán ha demostrado su capacidad de reducir las concentraciones sanguíneas de TTR. Tras 10-14 días de una dosis única de 300 microgramos, la concentración sérica media de TTR disminuyó en un 80%, y con la administración repetida cada 3 semanas, la reducción media de la TTR sérica al cabo de 9 y 18 meses de tratamiento se situó en el 83% y el 84%, respectivamente, manteniéndose estable en el tiempo con la administración continua.
Se debe recordar que la TTR es una proteína transportadora para la proteína de unión a retinol 4 (RBP4, por sus siglas en inglés), la transportadora principal de la vitamina A (retinol) en sangre. Por tanto, es previsible que la reducción en la TTR plasmática dé lugar a una reducción en los niveles plasmáticos de retinol a valores por debajo del límite inferior normal. Así, por ejemplo, a lo largo de 18 meses de tratamiento con patisirán se observó una reducción media del 45% de los niveles séricos de la proteína de unión al retinol, y del 62% en los de vitamina A (EMA, 2018a).
Inotersén es un 2′-O-2-metoxietil-fosforotioato de un oligonucleótido de cadena única formado por 20 unidades conectadas a través de 19 enlaces fosforotioato con una estructura de 5-10-5, es decir, está compuesto por 10 unidades de 2’-desoxirribonucleótidos centrales flanqueados por 5 unidades de 2’-O-(2-metoxietil)-ribonucleótidos en cada uno de los extremos (5’ y 3’). Todas las pirimidinas están metiladas en posición 5´, resultando la secuencia de nucleótidos4 siguiente: 5ʹ-UMe-CMe-UMe-UMe-G-G-T-T-A-CMe-A-T-G-A-A-A-UMe-CMe-CMe-CMe-3ʹ. La configuración absoluta de cada unidad de 2-desoxi-D-ribosa es (1R, 3S, 4R), y la de cada 2-O-(2-metoxietil)-D-ribosa es (1R, 2R, 3R, 4R). Como otros oligonucleótidos de fosforotioato, inotersén es una mezcla de 2n diastereoisómeros; con n= 19, es por tanto una mezcla de 219 (524.288) diastereoisómeros.
Tiene un nombre y estructura química complejos, se formula en solución estéril de la sal de sodio (nonadecasodio) y presenta una fórmula molecular de C230H299N69O121P19S19Na19, que se corresponde con un peso molecular de 7.600,8 Da. Inotersén fue seleccionado en un screening de entre más de 400 oligonucleóticos candidatos, y diseñado con unas modificaciones químicas que tienen como objetivo mejorar la estabilidad frente a la degradación mediada por nucleasas y reducir los posibles efectos secundarios asociados con la naturaleza polianiónica de los oligonucleótidos antisentido de fosforotioato. Se presenta como un sólido amorfo de color blanco a amarillo pálido, ampliamente soluble en agua y en tampones de fosfato a pH 7,5 y 8,5.
Por su parte, patisirán es un oligonucleótido de doble cadena, en el que cada una de las cadenas (sentido y antisentido) contiene 21 oligonucleótidos; 19 nucleótidos de cada una de las cadenas hibridan entre sí por complementariedad, formando una unión de 19 pares de bases que deja libres dos nucleótidos en los extremos terminales 3’ de cada cadena. Así, las cadenas sentido y antisentido de patisirán tienen, respectivamente, la siguiente estructura: 5ʹ-G-UMe-A-A-CMe-CMe-A-A-G-A-G-UMe-A-UMe-UMe-CMe-CMe-A-UMe-dT-dT-3ʹ y 3ʹ-dT-dT-C-A-UMe-U-G-C-U-U-C-U-C-A-UMe-A-A-G-G-U-A-5ʹ.
La fórmula molecular de patisirán, en forma de sal sódica, es C412H480N148O290P40Na40, siendo su peso molecular de 14.304 Da (7.304 Da de la cadena sentido y 7.100 Da de la cadena antisentido). Con respecto a su esteroquímica, todas las unidades de pentosa en patisirán aparecen en forma de D-ribosa. No se ha observado polimorfismo en la molécula. La sustancia activa es un polvo de color blanco-blanquecino, higroscópico y soluble en agua y soluciones salinas de tampón fosfato.
Hay que subrayar que patisirán se formula en nanopartículas de aproximadamente 60-100 nm de diámetro, con cuatro excipientes lipídicos especiales que, en conjunto, protegen al fármaco de la acción de endo- y exonucleasas en la circulación sistémica y facilitan su liberación en el órgano diana, el hígado (fuente primaria de la proteína TTR). Tras la infusión intravenosa de las nanopartículas, estas se revisten por la apolipoproteína E, lo cual facilita su unión a los receptores de lipoproteínas de baja densidad (LDL) en los hepatocitos, la fusión de los componentes lipídicos ionizables de la nanopartícula con la membrana del endosoma y la subsiguiente endocitosis; así, se libera el RNA de interferencia en el citoplasma.
La eficacia y la seguridad clínicas del inotersén por vía subcutánea han sido adecuadamente contrastadas en la indicación y dosis autorizada (284 mg una vez por semana) mediante un ensayo clínico randomizado de fase 2/3 (estudio NEURO-TTR), multicéntrico y multinacional (24 centros en 10 países), de grupos paralelos, doblemente ciego y controlado con placebo, de 15 meses de duración (66 semanas). Dicho estudio aleatorizó (2:1) un total de 173 pacientes adultos –172 recibieron al menos una dosis y 137 completaron el estudio– con amiloidosis hereditaria por transtiretina con polineuropatía en estadio 1 o 2 y una puntuación en la escala de deterioro NIS de ≥ 10 y ≤ 130 puntos, así como confirmación diagnóstica de depósitos amiloides en biopsia y documentación de la mutación de TTR por genotipado. Como criterios de exclusión se consideraron, entre otros, niveles anormalmente elevados de bilirrubina o transaminasas hepáticas, bajo recuento de plaquetas, niveles de retinol fuera del rango de normalidad o de la hormona TSH, hipertensión no controlada e infección por VIH o hepatitis B o C.
Las dos variables primarias empleadas para determinar la mejoría o la estabilización de los pacientes fueron el cambio medio desde el inicio a la semana 66 en la puntuación compuesta de la Escala de Deterioro por Neuropatía modificada con 7 pruebas (mNIS+7)5 y en la puntuación total en el cuestionario Norfolk de Calidad de Vida en neuropatía diabética6 (Norfolk QoL-DN, por sus siglas en inglés). Entre las variables secundarias de eficacia se consideraron la puntuación en el dominio de los síntomas (para pacientes en estadio 1) y de la funcionalidad física/afectación de fibras nerviosas largas (pacientes en estadio 2) del cuestionario Norfolk QoL-DN a la semana 66, así como evaluaciones nutricionales mediante el índice de masa corporal (IMC) o el IMC modificado (multiplicado por la albúmina sérica en g/l) en la semana 65.
Las características demográficas y de la enfermedad de los pacientes en el estado basal, que estuvieron bien balanceadas entre ambos grupos de tratamiento, se muestran junto con los principales resultados de eficacia en la Tabla 1 (Benson et al., 2018).
El análisis primario de los datos revela un beneficio estadísticamente significativo en favor del inotersén en la semana 66 en términos de las variables principales (escala mNIS+7 y cuestionario Norfolk QoL-DN). Si se consideran solo los datos de los 137 pacientes que completaron el estudio, las diferencias entre tratamientos son notablemente más marcadas entre el brazo de inotersén (N= 85) respecto a placebo (N= 52): se hallaron diferencias de medias de -19,73 puntos en la escala mNIS+7 (5,80 vs. 25,53 puntos de cambio a la semana 66; p < 0,001) y de -11,68 puntos según el cuestionario Norfolk QoL-DN (0,99 vs. 12,67 puntos de cambio a la semana 66; p < 0,001), a favor de inotersén. Además, los resultados en los componentes de mNIS+77 y los campos de las puntuaciones compuestas del Norfolk QoL-DN fueron coherentes con el análisis de las variables principales y mostraron un beneficio en neuropatías motoras, sensitivas y autonómicas.
Por otra parte, el análisis por subgrupos puso de manifiesto que la eficacia de inotersén –y su superioridad a placebo– es consistente a la semana 66 con independencia de características como la presencia de la mutación Val30Met, el estadio de la enfermedad, el tratamiento previo y la presencia de cardiopatía. En todas las comparaciones se halló significación estadística para ambas variables principales, con excepción de uno solo de los subgrupos en la puntuación total del cuestionario Norfolk QoL-DN (el de cardiomiopatía demostrada por ecocardiograma; p= 0,067). El efecto fue más pronunciado, si cabe, en el subgrupo de pacientes sin mutación Val30Met (diferencia de medias de -19,06 puntos en la escala mNIS+7 y de -9,87 puntos en el cuestionario Norfolk QoL-DN), en los pacientes con polineuropatía en estadio 2 (diferencia de -24,79 puntos en mNIS+7 y de -11,23 puntos en Norfolk QoL-DN) y en aquellos sin cardiomiopatía demostrada por ecocardiograma (diferencia de -18,79 puntos en mNIS+7 y de -11,67 puntos en Norfolk QoL-DN).
Con respecto a la seguridad clínica de inotersén, los datos que permitieron su caracterización también derivan del ensayo pivotal NEURO-TTR, complementados con aquellos del análisis intermedio del estudio ISIS 420915-CS3 (un fase 3 no aleatorizado, aún en marcha, que ha incluido a 134 pacientes que completaron el estudio pivotal, para ser tratados hasta un periodo total de 5 años). Se tienen datos de 1 año y 2 años de exposición a inotersén, respectivamente, en 112 y 69 pacientes, siendo el tratamiento más prolongado de 4,4 años en 2 pacientes; no obstante, los datos de seguridad a largo plazo son limitados para sacar conclusiones.
El perfil toxicológico del nuevo fármaco parece importante, pero aceptable. Prácticamente todos los pacientes tratados con inotersén, y también con placebo, reportaron algún evento adverso, muchos posiblemente relacionados con el propio avance de la enfermedad. Se produjeron eventos adversos relacionados con el tratamiento en el 78% de los pacientes tratados con inotersén (vs. 38% con placebo), siendo graves en el 28% de los casos (vs. 22% con placebo); así, la tasa de interrupción debida a eventos adversos también fue superior a placebo (14% vs. 3%), destacando los casos de trombocitopenia y glomerulonefritis. Por su frecuencia, sobresalen los siguientes eventos adversos asociados a inotersén: reacciones en el lugar de inyección (51%) –en general, autolimitadas y manejables, siendo eritema (31% vs. 0% con placebo), dolor (21% vs. 7%) y prurito (12% vs. 0%) los más comunes–, náuseas (31%), anemia (28%), cefalea (23%), pirexia (20% vs. 8%), edema periférico (19%), escalofríos (19% vs. 3%), vómitos (15% vs. 5%), trombocitopenia (13% vs. 2%) y recuento de plaquetas disminuido (11% vs. 0%). Como se ha sugerido, por su gravedad, hay que subrayar el riesgo de trombocitopenia (grave en el 7% de pacientes tratados con inotersén vs. 0% con placebo) y glomerulonefritis (3% vs. 2%).
En relación a su inmunogenicidad, hasta el 31% de los pacientes tratados con inotersén presentaron anticuerpos antifármaco al cabo de 15 meses de tratamiento, con un inicio tardío y bajo título; parece que en esos pacientes, las reacciones en la zona de inyección fueron más frecuentes, no observándose, en cambio, ninguna alteración de su eficacia o farmacocinética. Por último, aunque los eventos adversos oculares potencialmente relacionados con una deficiencia de vitamina A tuvieron una incidencia similar a placebo (en torno al 20%), existen incertidumbres sobre la conveniencia de suplementación con vitamina A durante el tratamiento, especialmente durante el embarazo.
Los datos de eficacia y seguridad clínicas de patisirán conducentes a su autorización por vía intravenosa proceden de un único ensayo pivotal aleatorizado de fase 3 (estudio APOLLO), multicéntrico y multinacional (52 centros en 21 países), de grupos paralelos, doblemente ciego y controlado con placebo, de 18 meses de duración. Incluyó un total de 225 pacientes adultos –193 completaron el estudio– con amiloidosis hereditaria por transtiretina con polineuropatía sintomática en estadio 1 o 2 y una puntuación en la escala de deterioro NIS de ≥ 5 y ≤ 130 puntos y en la escala de PND de ≤3b, así como documentación de mutación de TTR por genotipado.
Los pacientes fueron aleatorizados (2:1) a recibir bien una dosis de 300 µg/kg de patisirán (N= 148; 93% completaron tratamiento) o solución salina estéril como placebo (N= 77; 62% completaron tratamiento) mediante perfusión intravenosa una vez cada 3 semanas. Todos los pacientes recibieron premedicación con un corticoide, paracetamol y agentes bloqueantes H1 y H2 para reducir el riesgo de reacciones relacionadas con la perfusión. Se excluyeron pacientes con trasplante hepático previo (o previsión del mismo), neuropatía por otras causas, infección activa susceptible de tratamiento sistémico, cardiopatía no controlada, tratamiento concomitante con tafamidis o diflunisal o que hubieran participado en un ensayo clínico con oligonucleótidos antisentido (como inotersén) en los 3 meses previos a la aleatorización.
Las características demográficas de la población de estudio y las de la patología en el estado basal estuvieron bien equilibradas entre ambos brazos de tratamiento. En conjunto, la mediana de edad de los pacientes fue de 62 años, el 74% eran hombres y el 26% mujeres, el 46% tenía neuropatía en estadio 1 (neuropatía sensorial leve, motora y autónoma en miembros inferiores) y el 53% en estadio 2 (progresión moderada del deterioro a miembros inferiores, miembros superiores y tronco); se identificaron hasta 39 mutaciones diferentes de TTR, siendo la más frecuente la Val30Met8 (43% de pacientes, 10% de los cuales tenían síntomas de inicio antes de los 50 años); en torno a la mitad de los pacientes (53%) había recibido tratamiento previo con tafamidis, meglumina o diflunisal, y un porcentaje similar (56%) cumplía criterios de cardiopatía predefinida por amiloidosis.
La variable principal de eficacia del estudio fue el cambio medio desde el inicio hasta los 18 meses en la puntuación compuesta de la Escala de Deterioro por Neuropatía modificada con 7 pruebas (mNIS+7), considerándose como variable secundaria el cambio desde el inicio hasta los 18 meses en la puntuación total del cuestionario de calidad de vida de Norfolk-Neuropatía diabética (QoL-DN). Otras variables secundarias fueron, por ejemplo, la prueba de marcha de los 10 metros y el índice de masa corporal modificado.
Los resultados publicados (Adams et al., 2018) tras un análisis de los datos de la población por intención de tratar modificado demuestran un beneficio estadísticamente significativo para patisirán en la escala mNIS+7 en comparación con placebo (Tabla 2), también en todos los componentes individuales de dicha escala. Se observaron cambios destacables ya a los 9 meses de tratamiento (primera evaluación), cuando el tratamiento con patisirán produjo una diferencia de 16,0 puntos frente a placebo (cambio medio desde el inicio de -2,0 puntos vs. +14,0 puntos). Al final del estudio, la proporción de pacientes respondedores según el umbral de mNIS+7 (aquellos que mostraron una reducción en la puntuación desde el momento inicial) también fue significativamente superior en el brazo de tratamiento experimental (56,1% vs. 3,9% con placebo; p < 0,001).
El análisis por subgrupos reveló que el beneficio significativo con patirisirán frente a placebo en términos de la puntuación de la mNIS+7 y la QoL-DN de Norfolk fue similar en todos los subgrupos, incluidos edad, sexo, raza, región puntuación en la escala NIS, tipo de mutación TTR, tratamiento previo y estadio –gravedad– de la enfermedad. En pacientes con afectación cardíaca predefinida, en los que la eficacia del fármaco también fue consistente, las electrocardiografías demostraron una reducción notable en el grosor del tabique ventricular izquierdo (diferencia entre grupos: -0,9 mm; IC95% -1,7 a -0,2) y en la contractilidad longitudinal (diferencia: -1,37%; IC95% -2,5 a -0,3) en comparación con placebo; el marcador de afectación cardiaca propéptido natriurético de tipo B N-terminal (NT-proBNP) también redujo sus niveles notablemente en pacientes tratados con patisirán al mes 18, con una diferencia del 55% frente a placebo.
La seguridad clínica se definió por los datos procedentes de hasta 218 pacientes tratados con patisirán hasta un máximo de 3,7 años durante su desarrollo clínico, 148 de ellos en el estudio pivotal con una media de duración del tratamiento de 17,7 años. La práctica totalidad de pacientes tratados reportó algún evento adverso, severos solo en aproximadamente 1 de cada 3 pacientes (28% con patisirán vs. 36% en el grupo placebo); del global de eventos adversos, se relacionaron con el tratamiento en aproximadamente la mitad de pacientes (49% vs. 39% con placebo), de los cuales graves solo se describieron en el 2,7% (vs. 0% con placebo). Se observaron bajas tasas de interrupción (1,4% vs. 0% con placebo) o retirada (5% vs. 14%) del tratamiento experimental por motivos de seguridad (sobre todo fallo cardiaco o nefrotoxicidad, posiblemente asociadas con la progresión de la patología). Ningún caso de muerte (N= 7) se relacionó con el fármaco en estudio.
Las reacciones adversas más frecuentes en los pacientes tratados con patisirán fueron edema periférico (30% vs. 22%) y reacciones relacionadas con la perfusión (19% vs. 9%), entre las que destacaron el dolor de espalda, la rubefacción, el dolor abdominal o las náuseas; todas ellas fueron de gravedad leve-moderada y que se redujeron con el tiempo. No se observaron cambios destacables en análisis clínicos en relación con patisirán, sin cambios significativos en el recuento de plaquetas o en los niveles de marcadores de función renal o hepática. La incidencia de eventos adversos cardiacos fue muy similar en los pacientes tratados con patisirán o placebo. La inmunogenicidad del fármaco parece muy baja, pues solo se identificaron anticuerpos antifármaco en el 3,6% de los pacientes tratados, con títulos bajos y transitorios que no mostraron efecto en la eficacia o seguridad clínicas del tratamiento. Un estudio abierto de extensión, actualmente en marcha, aportará una mayor información sobre la seguridad de patisirán a largo plazo.
Inotersén es un nuevo oligonucleótido antisentido diseñado para unirse selectivamente a la región no traducida en 3’ (3’-UTR) del ARN mensajero (ARNm) de la transtiretina (TTR) humana, tanto de tipo mutante como normal o salvaje, y provoca, en consecuencia, su degradación a través de la escisión mediada por la RNAsa H1. Por su parte, patisirán es un ácido ribonucleico pequeño de interferencia bicatenario formulado en nanopartículas lipídicas que facilitan su estabilidad y llegada al hígado, donde se une específicamente a una secuencia conservada genéticamente en la región 3’-UTR no traducida del ARNm de la proteína TTR, también tanto en su forma mutante como salvaje, y a través de la interferencia de ARN (con mediación de la endonucleasa argonauta-2) produce la degradación catalítica de dicho ARNm.
Por tanto, aunque sus mecanismos difieren, ambos fármacos determinan una inhibición de la síntesis hepática de la proteína TTR y de su secreción a sangre, lo que se traduce en una reducción sustancial de los niveles séricos circulantes de la proteína y, con ello, en una mayor estabilización o aclaramiento de los depósitos de TTR amiloidótica y de las manifestaciones de la polineuropatía y cardiomiopatía. El efecto farmacológico de estos fármacos es novedoso en el sentido de que reducen los niveles tanto de la TTR de tipo mutado como salvaje, a diferencia del trasplante hepático ortotópico, que solo reduce la síntesis y niveles circulantes de proteína mutante (el órgano implantado seguirá sintetizando proteína TTR salvaje). Designados por la EMA como medicamentos huérfanos, ambos medicamentos han sido autorizados para el tratamiento de la polineuropatía en estadios 1 o 2 en pacientes adultos con amiloidosis familiar (o hereditaria) por transtiretina (ATTRh). No obstante, las pautas posológicas difieren: inotersén se administra por vía subcutánea una vez por semana y patisirán por vía intravenosa una vez cada 3 semanas9.
El ensayo clínico pivotal de inotersén, un estudio aleatorizado de fase 2/3 (NEURO-TTR) de grupos paralelos, doblemente ciego y controlado con placebo (N= 173), demostró que los pacientes asignados a recibir el fármaco durante 15 meses (N= 112) tenían una progresión notablemente menor de los síntomas neurológicos en comparación con placebo, con una diferencia estadísticamente significativa (p< 0,001) de casi 15 puntos en el cambio a la semana 66 en la escala mNIS+7 (aumento de 10,5 puntos vs. 25 puntos con placebo) y de 9 puntos en el cuestionario validado de calidad de vida Norfolk QoL-DN (aumento de 4 puntos vs. 13 puntos con placebo), consideradas como variables primarias. El beneficio, respaldado por los resultados de las variables secundarias, es mayor incluso si se consideran solo los pacientes que completaron el periodo de tratamiento, con diferencias medias de casi 20 y 12 puntos en mNIS+7 y en Norfolk QoL-DN, respectivamente. El análisis por subgrupos confirmó, además, que la superioridad de inotersén sobre placebo es consistente e independiente de factores como el tipo de mutación, gravedad del paciente, tratamiento previo o la presencia de cardiopatía: parece que los pacientes que se benefician en mayor medida son aquellos sin mutación Val30Met, en estadio 2 o sin cardiomiopatía.
El perfil toxicológico de inotersén está bien caracterizado a corto-medio plazo y es clínicamente manejable. Aunque los eventos adversos son frecuentes durante el tratamiento (en parte debido a la naturaleza y progresión de la enfermedad), en su mayoría son de severidad leve-moderada y autolimitados; los graves acontecen en el 28% de pacientes (vs. 22% con placebo), y la tasa de interrupción del tratamiento es superior a placebo (14% vs. 3%). Por su frecuencia, destacan las reacciones adversas en el lugar de inyección (51%) –sobre todo, eritema (31%), dolor (21%) y prurito (12%)–, náuseas (31%), anemia (28%), cefalea (23%) o pirexia (20%). Por su gravedad, sobresale el riesgo de trombocitopenia (13% vs. 2%, graves en el 7% vs. 0%) y de glomerulonefritis (3% vs. 2%), que hace conveniente la monitorización del recuento plaquetario y la tasa de filtración glomerular durante el tratamiento. Además, casi 1 de cada 3 pacientes presenta anticuerpos anti-fármaco que parecen relacionarse con mayor incidencia de reacciones en la zona de inyección. Pero habrá que esperar a conocer los resultados de un estudio de extensión en marcha para concluir sobre la seguridad de inotersén a largo plazo.
Por otra parte, la eficacia clínica de patisirán ha sido adecuadamente contrastada en un ensayo pivotal de fase 3 (APOLLO), también doble ciego y controlado con placebo (N= 225). Los pacientes adultos con polineuropatía en estadios 1 o 2 por ATTRh tratados con dicho fármaco durante año y medio mostraron una significativa mejoría o estabilización de las manifestaciones en comparación con placebo, revelada por una diferencia de 34 puntos en la escala mNIS+7 (cambio de -6 puntos vs. +28 puntos con placebo; p< 0,001) y apreciable ya desde los primeros 9 meses (diferencia de 16,0 puntos frente a placebo); al final del estudio, la proporción de pacientes respondedores según el umbral de mNIS+7 fue ampliamente mayor con patisirán (56% vs. 4%). Su superioridad sobre placebo se confirmó en los resultados de todas las variables secundarias, destacando su efecto, por ejemplo, sobre el cambio en la puntuación del cuestionario Norfolk QoL-DN (diferencia de -21 puntos a favor de patisirán), en la prueba de la marcha de 10 m o en el índice de masa corporal modificado; además, el análisis por subgrupos demostró que esa eficacia es independiente de factores como edad, sexo, raza, tipo de mutación TTR, tratamiento previo y estadio de la enfermedad.
En términos de seguridad, patisirán es un fármaco bien tolerado, con bajas tasas de interrupción (1,4% vs. 0% con placebo) o retirada (5% vs. 14%) del tratamiento, similares o inferiores a placebo. Aunque los eventos adversos se relacionaron con el tratamiento en la mitad de pacientes (49% vs. 39% con placebo), la gran mayoría fueron limitados en el tiempo y leves-moderados, con escasa incidencia de los graves (2,7% vs. 0%). Las reacciones adversas más frecuentes fueron edema periférico (30% vs. 22%) y reacciones relacionadas con la perfusión10 (19% vs. 9%), entre las que destacan: dolor de espalda, rubefacción, dolor abdominal o náuseas. Patisirán no afecta al recuento de plaquetas ni a la funcionalidad renal y hepática, y presenta una reducida inmunogenicidad (solo presentaron anticuerpos el 4% de los pacientes), que no altera su perfil beneficio-riesgo. Un estudio abierto de extensión, en desarrollo a día de hoy, aportará una mayor información sobre su seguridad a largo plazo.
Los dos ensayos clínicos mencionados tuvieron un diseño (tamaño, comparador, variables, etc.) y desarrollo que se consideran aceptables por la EMA en el contexto de una enfermedad rara como la ATTRh, que progresivamente conduce a la muerte, lo cual complica la replicación de resultados en un segundo estudio. A la vista de los hallazgos comentados, se considera que la eficacia de inotersén y patisirán en las manifestaciones clínicas y la calidad de vida de los pacientes con polineuropatía es clínicamente relevante, no así para los pacientes que solo presenten cardiomiopatía. Cabe destacar que han sido investigados solo en pacientes con polineuropatía en estadio 1 o 2 (que aún caminan), o sea, no han demostrado eficacia en los pacientes más graves en estadio 3 (alteración sensorio-motora generalizada, completamente dependientes, postrados en cama o silla de ruedas); pero a diferencia de tafamidis, sí serán útiles para el tratamiento de pacientes en estadio 2 (afectación de extremidades inferiores y manos, pueden todavía moverse con ayuda).
Aunque está por ver la influencia definitiva sobre la evolución global de la enfermedad a largo plazo (no hay datos sobre el impacto en supervivencia) y sus resultados clínicos puedan ser modestos, no cabe duda de que inotersén y patisirán aportan dos mecanismos de acción novedosos, con un efecto farmacológico similar (degradación del ARNm de TTR y menor síntesis proteica), e inauguran una nueva vía terapéutica en el manejo de la ATTRh, potencialmente útil en otros cuadros de amiloidosis asociados al depósito de fibrillas de proteínas anómalamente plegadas. Son, pues, dos nuevos tratamientos etiológicos modificadores del curso de la enfermedad cuya incorporación al mercado en una indicación en la que solo se disponía de una opción farmacológica (tafamidis) –útil además en una población más reducida de pacientes– es ciertamente relevante, especialmente si se considera que hasta dos tercios de los pacientes no son susceptibles de trasplante hepático (que se mantiene como el estándar terapéutico en muchos casos).
No se dispone aún de comparaciones clínicas directas entre las 3 opciones aprobadas para el tratamiento de la polineuropatía por ATTRh, lo que suele complicarse en enfermedades raras por los tamaños de muestra y costes asociados. Tampoco se han realizado comparaciones indirectas o meta-análisis que faciliten el posicionamiento de las distintas opciones; algunos autores incluso advierten de la dificultad de que éstos aporten una robustez aceptable (siempre limitada) dada la heterogeneidad entre los estudios pivotales de los tres fármacos que, aunque similares en diseño, tienen diferencias importantes en duración del tratamiento, criterios de selección de pacientes, características basales de los mismos o evaluación de la eficacia (Samjoo et al., 2020).
Teniendo en cuenta, por tanto, que no se puede aún concluir sobre la superioridad de uno u otro fármaco y por ahora se presentan como alternativas de tratamiento con distinto perfil de seguridad y pauta posológica, los resultados divulgados por una reciente revisión de la Cochrane (Magrinelli et al., 2020) y los datos comentados en este artículo podrían sugerir que inotersén y patisirán son más eficaces que tafamidis y pueden emplearse en un abanico más amplio de pacientes; han mostrado eficacia incluso en pacientes pre-tratados con tafamidis, por lo que su uso como 1ª línea condicionaría la posibilidad de emplear tratamientos posteriores en caso de progresión.
Si se compararan, de forma indirecta y no ajustada, los resultados numéricos de los dos nuevos fármacos, patisirán parece aportar un mayor beneficio clínico en términos de estabilización de las manifestaciones de la neuropatía periférica y de la calidad de vida de los pacientes. Por último, habiéndose aprobado previamente otros oligonucléotidos antisentido (con el mismo fundamento que inotersén) para el tratamiento de otras patologías, el hecho de que patisirán sea el primero de su clase –el primer ARN de interferencia que se autoriza para tratamiento en humanos– hace que este último represente una mayor innovación terapéutica.
El diagnóstico diferencial de las lesiones esplénicas es amplio, dada la gran variedad de formas de afectación del bazo, siendo, la gran mayoría de las lesiones de apariencia inespecífica, difíciles de comprobar histológicamente. Por tanto, se hace necesario considerar el contexto clínico del paciente en el que se detecta una lesión o una anomalía esplénica.
Existen diversas condiciones congénitas que involucran al bazo, y es de suma importancia conocerlas para evitar interpretaciones falsas, siendo un buen ejemplo de esto la presencia de hendiduras en la superficie esplénica, que pueden confundirse con laceraciones en el contexto de traumatismo abdominal. De igual modo, el bazo es un órgano típicamente involucrado de manera secundaria ante la presencia de diversas patologías sistémicas, pudiendo presentar lesiones en relación con la existencia de hemosiderosis, hipertensión portal o infecciones víricas o bacterianas; en este sentido, se debe considerar el bazo como uno de los órganos más frecuentemente involucrados en el contexto de infecciones diseminadas, máxime en pacientes inmunodeprimidos, aunque también puede verse afectado en el curso de infecciones localizadas en órganos vecinos, como el riñón izquierdo o el pulmón.
Por otro lado, las lesiones focales esplénicas primarias son, en su gran mayoría, benignas, y salvo los quistes o hemangiomas, suelen presentar un comportamiento poco específico. Las más frecuentes presentan unas características de imagen que permiten realizar un diagnóstico de aproximación. Una lesión focal esplénica aislada en un paciente con cáncer, pero sin enfermedad metastásica diseminada, no suele corresponderse a una metástasis.
El presente artículo pretende revisar los tipos de patologías que con más frecuencia afectan al bazo, considerando su etiología y principales características desde el prisma del diagnóstico clínico diferencial.
El bazo suele considerarse el “órgano olvidado”, probablemente porque no es necesario para vivir, y no es sustituido cuando se extirpa quirúrgicamente. Sin embargo, debido al uso generalizado de la ecografía, es muy frecuente en la práctica diaria y en diferentes contextos clínicos encontrar diversas lesiones en el bazo, de carácter inespecífico, que no tendrán comprobación histológica.
Existe una escasez de publicaciones científicas acerca de la patología esplénica, que se refleja en la escasa cantidad de artículos basados en la imagen del bazo, especialmente respecto a la detección y caracterización de la patología esplénica, si se compara, por ejemplo, con el hígado. Aunque muchas de las lesiones esplénicas comparten características radiológicas, la ecografía juega un importante papel en la caracterización de dichas lesiones, papel que se acentúa con el conocimiento preciso del contexto clínico del paciente.
El bazo es un órgano intraperitoneal localizado en el cuadrante superior izquierdo del abdomen, en el hipocondrio izquierdo. Forma parte del sistema retículo-endotelial, cumpliendo una función esencial en los procesos de filtración sanguínea y respuesta inmunitaria. Se caracteriza por una morfología lenticular y un contorno liso, siendo muy frecuente la presencia de bazos accesorios hasta en un 15% de los pacientes, así como hendiduras y lóbulos derivados de fallos en la fusión de las yemas celulares en la vida embrionaria.
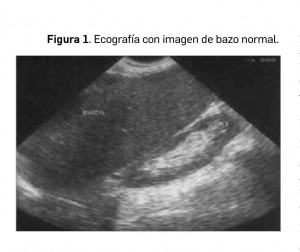
Su estructura histológica es también característica, formada por: la pulpa blanca, con folículos linfáticos y células reticuloendoteliales, y la pulpa roja, con las sinusoides vasculares. La proporción de ambas varía con la edad, aumentando la pulpa blanca con la edad debido al acúmulo en la exposición a los antígenos. En ecografía, el bazo presenta una ecoestructura homogénea, siendo ligeramente hiperecogénico respecto a la corteza renal y al hígado (Figura 1).
El tamaño del bazo varía con la altura y el peso corporal, alcanzando el volumen máximo en los adultos jóvenes, y disminuyendo con la edad. No hay acuerdo acerca del tamaño considerado normal, aunque se establece un consenso de 13 cm de diámetro craneocaudal como límite alto de la normalidad. El embarazo es un estado especial en el que el bazo aumenta de tamaño, de forma leve o moderada, de manera fisiológica, debido al aumento del volumen sanguíneo materno hasta en un 45%; no se conoce con exactitud cuándo debe el bazo volver a su estado normal tras el parto y el puerperio.
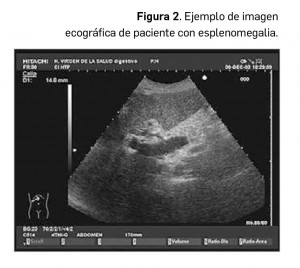
En nuestro medio, la hipertensión portal es la causa más frecuente de aumento de tamaño del bazo o esplenomegalia (Figura 2), recomendándose un estudio exhaustivo para descartar la existencia de circulación colateral y signos de cirrosis hepática, en casos de detección casual de esplenomegalia en cualquier estudio rutinario.
Como segunda causa más frecuente de esplenomegalia, se consideran las enfermedades hematológicas, concretamente linfoma, leucemia mieloide crónica, policitemia vera, hemoglobinopatías, leucemia linfática crónica, mielofibrosis y esferocitosis. Y, en tercer lugar, también puede aparecer un aumento de tamaño esplénico por enfermedades infecciosas, dependiendo del contexto geográfico: la infección por el virus de la inmunodeficiencia humana (VIH), la mononucleosis infecciosa y la leishmaniasis con las más frecuentes en nuestro medio, mientras que, en pacientes procedentes del continente africano, las más prevalentes son la malaria y la esquistosomiasis.
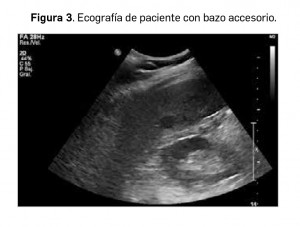
Se trata de la anomalía congénita esplénica más frecuente, encontrada en hasta el 10-30% de las autopsias. Se produce como consecuencia del fallo de fusión de los grupos de células mesenquimales que dan lugar al bazo en la vida fetal. Es importante reflejarlo en el informe clínico del paciente a fin de no confundirlo con la presencia de una adenopatía o de un implante nodular tumoral en los sucesivos estudios. De igual modo, es importante la extirpación completa en casos de esplenectomía programada, para evitar recidiva de la enfermedad de base. El lugar más común de localización del bazo accesorio es el hilio esplénico (Figura 3).
Cabe destacar que el bazo accesorio sufre las mismas alteraciones que el bazo en la patología esplénica, como la congestión esplénica secundaria a hipertensión portal, el depósito de hierro en la hemosiderosis o en los síndromes linfoproliferativos, o la hipertrofia en caso de esplenectomía.
No presentan significación clínica. Habitualmente los lóbulos fetales de tejido esplénico desaparecen en la vida adulta, aunque pueden persistir dando lugar a la presencia de lóbulos en el bazo adulto. Las hendiduras esplénicas son remanentes de los surcos que separan los diferentes lóbulos fetales, pudiendo presentar hasta 2-3 cm de profundidad, y ocasionalmente, confundirse con laceraciones esplénicas en pacientes que han sufrido traumatismo abdominal cerrado.
Se trata de un estado de hipermovilidad del bazo derivado de la excesiva laxitud de los ligamentos de sostén (ligamento esplenorrenal y ligamento gastroesplénico) que anclan el órgano a la pared abdominal y a las estructuras vecinas, o bien del desarrollo anómalo de éstos; ello permite que el bazo se localice no solo en el hipocondrio izquierdo sino también en el abdomen inferior e incluso en la pelvis. Es importante reconocer esta condición, dado que la complicación más importante es la torsión del pedículo, con el consiguiente compromiso vascular e infarto esplénico. El tratamiento es la esplenopexia preventiva, o la esplenectomía urgente en caso de torsión del pedículo e infarto.
La apariencia ecográfica de las lesiones focales es altamente inespecífica, con gran solapamiento entre diferentes entidades, no siendo posible, en la mayoría de los casos, llegar a un diagnóstico o acotar el diagnóstico diferencial. Por tanto, es de crucial importancia la información clínica del paciente con lesiones focales esplénicas en el momento de la realización de la exploración. Por este motivo, se deben valorar las lesiones focales esplénicas según los diferentes contextos clínicos:
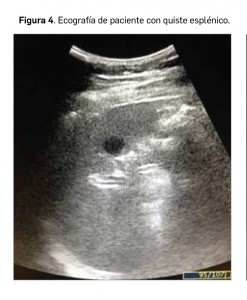
Son lesiones no neoplásicas que constituyen las lesiones focales esplénicas más frecuentes en la población pediátrica. Se pueden considerar como: quistes verdaderos, aquellos que están tapizados de epitelio y con origen mayoritariamente congénito o parasitario, o pseudoquistes o “falsos quistes”, cuando el epitelio está ausente y normalmente son adquiridos como consecuencia de degeneración quística de hematomas esplénicos producidos por un traumatismo previo.
En ecografía (Figura 4), la apariencia es la típica de las lesiones quísticas, es decir, lesiones bien delimitadas, de contornos lisos y contenido líquido, siendo más frecuente la presencia de septos o tabiques en los quistes congénitos, a la par que es más frecuente la presencia de calcificaciones en los seudoquistes o quistes adquiridos. Otro tipo de lesiones quísticas en el bazo son menos frecuentes, destacando entre ellas el linfangioma quístico, que se caracteriza por la presencia de múltiples lesiones con morfología “en panal de abeja”.
A pesar de la alta prevalencia de la hidatidosis hepática en nuestro medio, el quiste hidatídico esplénico (quiste verdadero), sin ser excepcional, es muy poco frecuente, sobre todo en ausencia de enfermedad hepática o peritoneal.
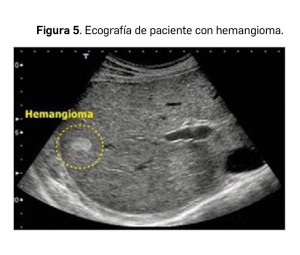
El hemangioma es la lesión primaria benigna esplénica más frecuente. Su apariencia ecográfica (Figura 5) es similar a los hemangiomas hepáticos, siendo hiperecogénicos y de menor tamaño.
Es una lesión benigna poco frecuente, compuesta de elementos malformados de la pulpa roja, de la pulpa blanca, o más frecuentemente, de una combinación de ambas. Típicamente, aparece como una lesión única, solitaria, de tamaño variable, con una media de 5 cm, habiéndose descrito hamartomas de hasta de 20 cm. Su apariencia radiológica es heterogénea, en parte debido a la presencia de focos de degeneración quística, calcificación o fibrosis. En ecografía (Figura 6, imagen izquierda), es frecuente encontrarlos como lesiones hiperecogénicas bien definidas con señal Doppler color muy marcada.
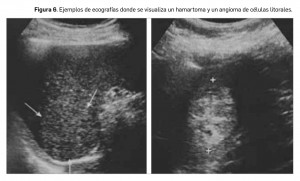
Se trata de un tumor exclusivamente esplénico, de carácter vascular y muy poco frecuente, con origen en las células de la pulpa roja del bazo, concretamente en las células que tapizan los sinusoides, también llamadas “células litorales”. Fue descrito por primera vez en 1991. Habitualmente se presenta como múltiples lesiones focales esplénicas (Figura 6, imagen derecha), lo que plantea el diagnóstico diferencial con hemangiomas, linfangiomas, metástasis, sarcoidosis, microabscesos esplénicos y linfoma. Pero también ha sido descrito como una lesión focal única. El diagnóstico se realiza tras la extirpación quirúrgica y el estudio histológico, ya que las características ecográficas son inespecíficas.
Es relativamente frecuente encontrar de manera ocasional pequeñas calcificaciones en el bazo en estudios ecográficos. En la mayoría de las ocasiones, corresponden a granulomas resueltos y calcificados, secundarios a enfermedad granulomatosa, típicamente por tuberculosis en nuestro medio, o histoplasmosis en otros países. Aparecen como focos de calcificación puntiformes y dispersos en el parénquima esplénico, que suelen asociarse a calcificaciones también en el hígado y en los ganglios linfáticos. Es importante recordar que no es necesario realizar más exploraciones ante la visualización de granulomas esplénicos.
Como se ha comentado en el apartado de lesiones quísticas, es frecuente la calcificación de lesiones esplénicas postraumáticas en su evolución desde el hematoma agudo inicial, hasta degeneración quística parcial o totalmente calcificada. De la misma forma, los infartos esplénicos se pueden calcificar en su proceso evolutivo hacia la cronicidad.
Mención especial merece esta patología hematológica como causa de calcificaciones esplénicas. Clásicamente, en estos enfermos, el bazo se destruye en la primera década de la vida debido a crisis vasooclusivas o crisis isquémicas, en un proceso denominado “autoesplenectomía”, por el que el bazo se va transformando en un órgano pequeño y fibrótico, habitualmente calcificado, ocurriendo este proceso en todos los adultos homocigóticos y siendo la característica principal de esta enfermedad.
Sin embargo, infartos silentes o subclínicos también son capaces de producir la total calcificación del bazo, ante lo cual deberá iniciarse el estudio genético del paciente para detectar a los pacientes heterocigotos con dicho rasgo genético, sin historia clínica de infartos esplénicos.
No se trata de verdaderas calcificaciones. Son depósitos de hemosiderina dispersos en el parénquima esplénico, típicos de pacientes con hipertensión portal. En ecografía, es frecuente encontrarlos en pacientes con cirrosis sometidos a cribado semestral de hepatocarcinoma, visualizando un punteado hiperecogénico sin sombra posterior, que en resonancia magnética (RM) se comportan como focos puntiformes de hiposeñal en todas las secuencias, más llamativo en secuencias con mayor susceptibilidad magnética, como las secuencias T2.
El bazo puede estar involucrado en las enfermedades infecciosas, bien como foco único de infección o bien formando parte de la afectación generalizada de la enfermedad. Una gran variedad de infecciones puede diseminarse hematológicamente y producir abscesos esplénicos o microabscesos. No obstante, la infección esplénica aislada es rara, siendo lo habitual la presencia de infección en el hígado y en otros órganos al mismo tiempo.
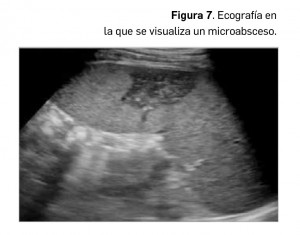
El contexto clínico suele incluir: fiebre, mal estado general y dolor en el hipocondrio izquierdo, no siendo el hallazgo casual de esta afección en absoluto frecuente. Los pacientes inmunodeprimidos son proclives al desarrollo de afectación esplénica infecciosa, habitualmente por diseminaciones fúngicas, siendo la más frecuente la candidiasis generalizada, seguida en frecuencia por la aspergilosis y la criptococosis. Los pacientes con tuberculosis miliar diseminada también suelen presentar afectación esplénica múltiple, habitualmente en forma de microabscesos. Los microabscesos esplénicos se manifiestan como lesiones milimétricas de baja ecogenicidad (Figura 7), dispersas en el parénquima esplénico, acompañándose de esplenomegalia.
En este contexto, lo más frecuente es la afectación hepática y esplénica concomitante. Además, en el caso de la tuberculosis diseminada, suelen coexistir la presencia de adenopatías y líquido libre abdominal por afectación peritoneal. Por otro lado, los abscesos esplénicos únicos, aunque poco frecuentes, pueden aparecer en pacientes con infecciones bacterianas. La endocarditis es la causa más frecuente de abscesos esplénicos como fuente de diseminación infecciosa. Asimismo, la extensión al bazo de un proceso infeccioso en vecindad, o la sobreinfección de un infarto o lesión traumática son otros de los mecanismos de producción de abscesos esplénicos. Pueden aparecer secundariamente a pielonefritis o abscesos renales izquierdos o en contigüidad a empiema pleural y/o neumonía basal izquierda.
Ante un paciente con dolor en el flanco izquierdo, debemos sospecha de la posible presencia de:
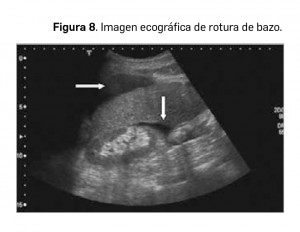
El bazo es el órgano abdominal más frecuentemente lesionado tras un traumatismo abdominal cerrado, tanto leve como grave. Las causas más frecuentes engloban desde la caída accidental hasta el traumatismo de alta energía por accidente de tráfico, así como la biopsia esplénica o la rotura espontánea (Figura 8) en pacientes con esplenomegalia o terapia anticoagulante. Las lesiones esplénicas traumáticas son muy variadas, pudiendo variar desde contusiones leves, que se tratan de manera conservadora, hasta estallidos que requieren una cirugía urgente.
No se trata de una lesión esplénica aguda ni se detecta en el contexto de dolor en el hipocondrio izquierdo, sino que es la consecuencia de una rotura esplénica donde se ha realizado posteriormente una esplenectomía (Figura 9). Se define como la presencia de tejido esplénico ectópico, habitualmente de morfología nodular, disperso en la cavidad abdominal, más frecuentemente en la superficie de la serosa del intestino delgado, peritoneo, mesenterio y diafragma. No presenta síntomas relacionados, por lo que el diagnóstico es casual en la mayoría de ocasiones.
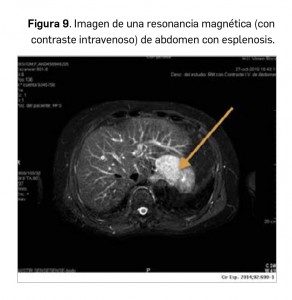
La presencia de focos de esplenosis en el hipocondrio izquierdo de localización subfrénica, en la teórica localización del bazo previamente extirpado, es lo más habitual. No obstante, también pueden depositarse en la superficie hepática, por lo que no es extraño la confusión en el diagnóstico de focos de esplenosis con verdaderas lesiones hepáticas (como los hemangiomas), e incluso, en el contexto de cirrosis hepática o malignidad, pueden llegar a ser interpretados erróneamente como un hepatocarcinoma o zonas de metástasis.
De la misma manera, cuando la esplenosis es muy florida debido a un antecedente de estallido esplénico, y se dispone de manera multinodular y dispersa en la cavidad abdominal, es posible el diagnóstico erróneo de implantes tumorales peritoneales, siendo un reto diagnóstico, sobre todo si se está en un contexto neoplásico. En estos casos, y cuando sea indispensable establecer con precisión el diagnóstico diferencial entre las dos entidades, la gammagrafía con hematíes marcados con tecnecio-99, junto con el antecedente de rotura esplénica, proporcionarán el diagnóstico de confirmación de esplenosis con alta especificidad.
El infarto esplénico es relativamente frecuente en pacientes con enfermedades hematológicas o patología tromboembólica de base. En un alto porcentaje de enfermos, los pequeños infartos cursan de modo asintomático y son descubiertos de manera incidental al realizar una ecografía por otros motivos. Su apariencia más frecuente es la presencia de áreas hipoecoicas de morfología triangular y base periférica. En ocasiones, coexisten con infartos en otras localizaciones como el riñón, en casos de patología tromboembólica de base, presentando en estos casos una sintomatología aguda más importante. La afectación global esplénica por infartos de repetición es característico de la anemia de células falciformes.
Este escenario es muy frecuente en la práctica clínica diaria, siendo extremadamente habitual la detección de lesiones focales en el bazo de pacientes con neoplasia de base a los que se les realizan estudios en el contexto de la estadificación inicial del cáncer o en los controles de vigilancia posterior o de evaluación de cualquiera de los tratamientos. En estos casos, se debe ser cauto en la caracterización de dichas lesiones, que, por otro lado, suelen ser de pequeño tamaño y con apariencia inespecífica.
Conviene recordar que el bazo ocupa el décimo lugar en frecuencia en cuanto a la presencia de metástasis, siendo raras las metástasis aisladas en el bazo; por tanto, en el caso de encontrar una lesión focal esplénica en un paciente con una neoplasia conocida, la probabilidad de que corresponda a una lesión primaria es mucho mayor de que corresponda a una lesión metastásica. En este contexto, además, habrá que valorar la presencia de posibles metástasis en otras localizaciones, como el pulmón, el hígado, los ganglios linfáticos o el peritoneo, ya que la afectación metastásica del bazo (Figura 10) es más probable cuanto más diseminada o mayor carga tumoral tenga la enfermedad de base.
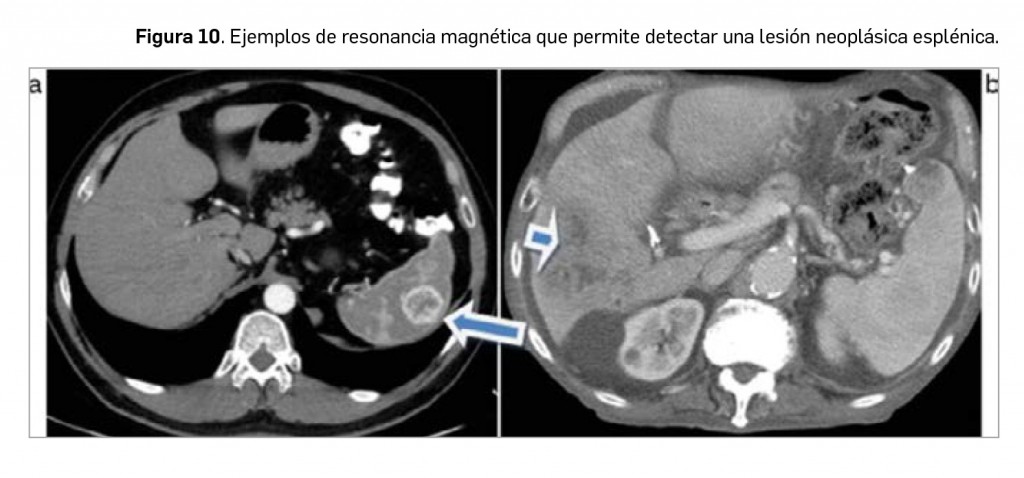
Por tanto, se puede afirmar que una lesión focal esplénica aislada en un paciente con cáncer, pero sin enfermedad metastásica diseminada, no suele corresponder a una metástasis. Las neoplasias que más frecuentemente producen metástasis en el bazo son el cáncer de pulmón, el de mama y el melanoma maligno.
Los mecanismos de desarrollo de las metástasis esplénicas pueden ser los siguientes:
No obstante, el bazo se ve afectado por tejido neoplásico debido a la presencia de implantes tumorales en la superficie del órgano, como ocurre en los casos de carcinoma ovárico, por neoplasias del tracto gastrointestinal y por la neoplasia de páncreas diseminada. De igual modo, es bastante habitual que el bazo se afecte por extensión directa de neoplasias en vecindad, siendo las más frecuentes, obviamente, el cáncer de la cola del páncreas, el cáncer gástrico y el carcinoma dependiente del riñón izquierdo.
También pueden identificarse otros dos trastornos neoplásicos en el hígado:
La causa de afectación neoplásica más frecuente del bazo es el linfoma, en cuanto que el bazo participa del contexto tumoral. El linfoma primario esplénico es una entidad poco frecuente (explica únicamente el 1% del conjunto de linfomas), y habitualmente del tipo no Hodgkin variante de células grandes; se manifiesta como una lesión focal esplénica con presencia de adenopatías en el hilio esplénico.
En cambio, la afectación secundaria del bazo es muy frecuente en el caso de linfomas, tanto Hodgkin como no Hodgkin. Puede aparecer de diferentes formas, aunque la más frecuente es la presencia de esplenomegalia con afectación nodular miliar. La existencia de lesiones focales múltiples, o incluso el hallazgo de una única lesión focal (como en el caso del linfoma de células grandes) es posible (Figura 11).
Es la neoplasia maligna primaria del bazo más frecuente, aunque es extremadamente rara. Es propia de pacientes en la sexta-séptima décadas de la vida, y más frecuente en mujeres. Tiene un origen vascular en las células del endotelio de los sinusoides esplénicos y es altamente agresiva, presentando una elevada mortalidad. En ecografía (Figura 12), se manifiesta como lesiones heteroecoicas, con gran componente de necrosis y/o hemorragia, que en el momento del diagnóstico suele estar diseminada con presencia de metástasis hepáticas muy vascularizadas.
La esclerosis lateral amiotrófica o ELA es una enfermedad neurodegenerativa que afecta a las motoneuronas de cerebro y médula espinal y produce una debilidad y paralización muscular progresiva del individuo, sin que en principio se afecten otras funciones del sistema nervioso diferentes a las motoras. Conduce a la muerte –por insuficiencia respiratoria– en un periodo de tiempo variable tras el diagnóstico, pero generalmente corto (3-5 años). Se trata de la tercera enfermedad neurodegenerativa en incidencia, tras la demencia y la enfermedad de Parkinson, y suele debutar principalmente entre los 50 y 70 años de edad. Cada año se diagnostican unos 2-5 casos por cada 100.000 habitantes a nivel mundial, afectando tanto a hombres como a mujeres (ligeramente más frecuente en los primeros). A pesar de su incidencia elevada, tiene una prevalencia baja por su elevada mortalidad: se estima que en el mundo hay medio millón de personas con la enfermedad. En España, la incidencia es similar al resto de países desarrollados, y anualmente se detectan 1,4 casos por 100.000 habitantes, lo que se traduce en unos 3 nuevos pacientes diagnosticados cada día y un total de aproximadamente 4.000 personas que viven con la enfermedad.
Si bien se han relacionado con la ELA más de 30 genes, se trata de una patología de causa desconocida en la que se ha descrito la participación de diversos factores ambientales y mecanismos fisiopatológicos de muerte neuronal. Pese a su escasa prevalencia, constituye un problema de salud con un alto impacto sociosanitario y económico, en ocasiones ignorado en la sociedad, por su gravedad y por el sufrimiento que supone para los pacientes y sus familias: su gran capacidad invalidante, la necesidad de cuidados permanentes y cambiantes, la gravedad de las complicaciones y los problemas emocionales y psicológicos que genera son aspectos diferenciales que requieren respuestas ágiles, coordinadas y accesibles para el enfermo y su entorno familiar.
En ausencia de cura y de opciones que modifiquen eficazmente el curso de la patología, el tratamiento actualmente disponible, que engloba farmacoterapia y medidas no farmacológicas, es paliativo y multidisciplinar, y se enfoca fundamentalmente al manejo de los síntomas (espasticidad, calambres musculares, alteraciones del sueño y problemas de salivación, entre otros) y la mejora de la calidad de vida. En todo caso, resulta fundamental el impulso de la investigación biomédica que permita a los pacientes alumbrar esperanza en el futuro. El presente artículo revisa ampliamente el conocimiento sobre la etiopatogenia, la epidemiología, las manifestaciones clínicas y el tratamiento de la ELA, centrando el foco en el avance –pasado y reciente– de la investigación científica. Se pone en valor, finalmente, el importante papel asistencial que el profesional farmacéutico puede ejercer para con el paciente con ELA y sus cuidadores, en términos de educación y orientación sociosanitarias, detección precoz y optimización de la farmacoterapia.
La esclerosis lateral amiotrófica (ELA) es una enfermedad neurodegenerativa caracterizada por una parálisis muscular progresiva que refleja una degeneración de las neuronas motoras en el sistema nervioso central y, concretamente, en la corteza motora primaria, tracto corticoespinal, tronco encefálico y médula espinal. En esta patología se produce una parálisis de los músculos voluntarios que se va extendiendo desde un punto de afectación inicial hacia regiones adyacentes, y de forma continuada y progresiva va afectando a la movilidad, al habla, la deglución o la respiración; sin embargo, la movilidad ocular, la sensibilidad cutánea, el control de los esfínteres y las funciones mentales se mantienen intactos en la mayor parte de los casos (Barrera et al., 2017).
De forma general, todas las enfermedades neurodegenerativas se caracterizan por una disminución en el número de células en determinadas poblaciones neuronales y el consiguiente deterioro funcional de las partes afectadas. Se han identificado más de 100 tipos de enfermedades neurodegenerativas, que comparten el curso progresivo de los síntomas (por la desintegración paulatina de una o varias partes del sistema nervioso), sin remisiones, y un inicio insidioso. Puesto que la mayoría de ellas son de etiología desconocida, se clasifican según las manifestaciones clínicas, distinguiendo entre: las que cursan con un síndrome demencial (enfermedad de Alzheimer y otras demencias neurodegenerativas), las que se manifiestan con trastornos del movimiento y la postura (síndromes parkinsonianos, como la propia enfermedad de Parkinson, y síndromes discinéticos, como la enfermedad de Huntington, distonías, etc.), los síndromes espinocerebelosos (las diversas formas de ataxia y la paraplejia espástica hereditaria), las polineuropatías periféricas y las enfermedades de las neuronas motoras o motoneuronas, en las que la debilidad y atrofia muscular son la manifestaciones fundamentales.
La ELA es la enfermedad de neurona motora1 más frecuente en el adulto, y también la más grave. La expresión “esclerosis lateral” se refiere a la pérdida de fibras nerviosas acompañada de una cicatrización glial o esclerosis en la región lateral de la médula espinal, y amiotrófica –término que procede del griego (a: partícula privativa o negativa, mio: músculo, y trófico: nutrición)–, a la atrofia muscular por afectación de las neuronas motoras inferiores, acompañado de debilidad y fasciculaciones.
Fue descrita en términos clínicos por primera vez por Charles Bell en 1830, aunque fue el médico francés Jean-Martin Charcot quien en 1869 realizó el primer estudio sistemático de la ELA, describiendo las características clínico‐patológicas de la enfermedad de forma muy similar a como se conocen actualmente. Además de conocerse como enfermedad de Charcot, sobre todo en Francia, también se la denomina enfermedad de Lou Gehrig en EE.UU., en honor a Henry Louis Gehrig, quien es allí considerado como el mejor jugador del baseball del siglo XX y murió en Nueva York a los 38 años de edad, apenas 2 años después de habérsele diagnosticado ELA. Más conocido en todo el mundo es el caso de Stephen Hawking, el célebre físico y divulgador científico británico que murió por ELA en 2018 y cuyo estado físico se fue agravando paulatinamente hasta dejarlo prácticamente paralizado, forzándole a comunicarse a través de un dispositivo informatizado generador de voz; le fue diagnosticada una ELA en 1963, con apenas 21 años de edad, momento en el que se le dio un pronóstico desfavorable (2 a 3 años de vida), pero su actitud fue, sin duda, un ejemplo de lucha y una clara manifestación de que la ELA no limita en muchos casos la extraordinaria lucidez mental (Cuéllar, 2013).
Según se ha sugerido anteriormente, todavía no se conocen los mecanismos causantes de la ELA y su desarrollo, ni su origen concreto. Se cree que el origen de la ELA es multifactorial, involucrando a un número variado de tipos celulares y diversos mecanismos que conllevan una reducción funcional de la actividad motora.
Además de la edad avanzada (considerado factor de riesgo común de todas las enfermedades neurodegenerativas) y el sexo (masculino), los únicos factores de riesgo establecidos son los factores genéticos. Otros, como la mayor actividad física, se encuentran en estudio como posibles factores de riesgo (Lisle et al., 2015).
Aproximadamente el 90% de casos de ELA ocurren de forma esporádica, sin historia familiar ni factores claramente asociados, atribuyéndose a interacciones entre los diversos factores de riesgo, esto es, a la interacción genética-medio ambiente, incluyendo el propio estilo de vida. Por ejemplo, en algunas islas del Pacífico Sur (la isla de Guam, en las Marianas, así como en Nueva Guinea y en la península de Kii en Japón) la incidencia de la ELA aumenta en un orden (10 veces) apareciendo casos con comorbilidad de demencia y parkinsonismo. En estos casos se ha estudiado el medioambiente y la alimentación, relacionando la aparición de la enfermedad con el consumo del fruto de una planta originaria llamada cica. Aunque los familiares de las personas con ELA esporádica corren un mayor riesgo de sufrir la enfermedad, el riesgo en general es muy bajo y la mayoría no la desarrolla.
El restante 10% de los casos se consideran casos familiares, normalmente resultado de mutaciones genéticas de herencia autosómica dominante (aunque hay algunos casos de herencia autosómica recesiva o ligada al sexo), lo que supone que es suficiente para desarrollar la enfermedad el que uno solo de los padres porte la mutación responsable de la enfermedad. Los casos de ELA familiar suelen ocurrir en personas que poseen o han tenido un familiar de primer o segundo grado con ELA o con demencia frontotemporal (los primos son considerados hallazgos casuales, quedando dentro de la definición de ELA esporádica).
Si bien de hasta 1 de cada 4 casos de ELA familiar aún no ha sido elucidada su causa genética o molecular, hasta la fecha se han encontrado ya mutaciones relacionadas con ELA familiar en más de 30 genes. Cerca del 25-40% de todos los casos familiares, y hasta el 10% de casos esporádicos, presentan un defecto –expansión repetida del hexanucleótido CCCCGG (del orden de cientos de veces, mientras que en la población general se repite entre 2 y 25 veces)– en un gen conocido como “marco 72 de lectura abierta en el cromosoma 9” o C9orf72, por sus siglas en inglés. Algunas personas portadoras de esta mutación pueden mostrar signos tanto de afectación de las neuronas motoras como síntomas de demencia, pues la misma mutación puede estar asociada con la atrofia de los lóbulos frontal y temporal del cerebro que origina la demencia frontotemporal (ELA-DFT), de la que es también la causa en un 40% de los casos.
En otro 15-20% de los casos familiares (un 1-2% de todas las formas de ELA) están implicadas mutaciones en el gen codificante de la síntesis de la enzima cobre-zinc superóxido dismutasa 1 (SOD1)2, enzima citosólica que interviene en la eliminación de radicales libres intracelulares, asociados con la patogenia de la ELA y de otras muchas enfermedades. Aún se desconoce la causa que origina la enfermedad en las familias con estas mutaciones, si bien se cree que en el proceso se produce la acumulación de proteínas aberrantes en el interior de la neurona motora de forma específica, entre ellas el propio enzima SOD1.
El hallazgo de estas y otras mutaciones relacionadas con la ELA de forma menos frecuente (por ejemplo, en los genes TARDBP, FUS, VCP o PNF1), que no obstante también se identificado en algunos casos esporádicos) sugieren que los defectos en el procesamiento de las moléculas de ARN y en el proceso natural de reciclaje de proteínas –por el cual las proteínas que funcionan mal se descomponen y se utilizan para construir nuevas– estarían implicadas en la degeneración de las motoneuronas, las cuales, además, también podrían presentar defectos en su estructura y forma, así como una susceptibilidad incrementada a las toxinas ambientales (NINDS, 2017).
Todavía hoy carecemos de un modelo concreto que justifique la muerte selectiva de las neuronas motoras en la ELA (en el asta ventral de la médula espinal, los núcleos motores del tronco encefálico y las neuronas motoras superiores de la corteza cerebral), aunque se ha observado la presencia de inclusiones intraneurales inmunorreactivas de ubiquitina en las neuronas motoras superiores y de agregados citosólicos anormales de TDP‐43 (TAR DNA‐binding protein 43, codificada por el gen TARDBP y normalmente de ubicación nuclear) en las neuronas motoras inferiores, incluso en pacientes que también presentan mutación de C9orf72.
Lo que sí resulta evidente es que un cerebro de un paciente con ELA tiene, como principales cambios microscópicos, pérdida de neuronas y axones (Saveri et al., 2015). Con respecto al mecanismo específico de la muerte neuronal, se ha postulado que puede ser la consecuencia de un proceso de excitotoxicidad mediada por ácido glutámico. El glutamato es un neurotransmisor excitatorio que desempeña un papel decisivo en el funcionamiento neurológico normal como respuesta rápida a estímulos y está estrechamente implicado en los procesos de cognición, memoria, movimiento y sensibilidad, pero a niveles excesivos, es tóxico (excitotóxico) y se ha demostrado que puede causar daño neurológico e inducir la apoptosis neuronal. Su liberación en grandes cantidades provoca hipertactividad glutamatérgica, que se asocia a la acción sobre sus receptores específicos en la membrana neuronal (receptores inotrópicos3 AMPA y NMDA, y receptores metabotrópicos), cuya activación pone en marcha mecanismos que facilitan la entrada masiva de iones Ca2+ desde el espacio extracelular, y provoca la activación de mecanismos enzimáticos intracelulares conducentes a la apopotosis neuronal. Según este planteamiento, la muerte neuronal selectiva se debería a que las motoneuronas tienen una menor capacidad de contrarrestar el fuerte incremento de las concentraciones intracelulares de Ca2+, por la menor expresión de proteínas que se unen a éste y facilitan su salida celular.
Otros posibles mecanismos involucrados en la patogénesis de la ELA que se han ido dilucidando gracias a los modelos animales y celulares de la enfermedad –ratones transgénicos que sobreexpresan el gen humano mutado SOD1 y otros modelos de expresión de genes últimamente relacionados con ELA familiar en animales transgénicos y en células pluripotenciales inducidas–, compatibles e incluso complementarios a los anteriores, han sido el estrés oxidativo (resultante de un desequilibrio entre la generación de especies reactivas de oxígeno y su eliminación y/o de la capacidad del sistema biológico para el daño producido por ellas), la disfunción mitocondrial, alteraciones en el citoesqueleto y en el transporte axoplásmico y los fenómenos de neuroinflamación y autoinmunidad.
En este sentido, la presencia de inclusiones intraneurales de ubiquitina en las motoneuronas superiores de los pacientes con ELA es coherente con el modelo de la excitotoxicidad, teniendo en cuenta que esta proteína, presente en la mayoría de los tejidos tiene, entre sus múltiples funciones, la de dirigir el reciclaje de proteínas uniéndose a ellas y marcándolas para su destrucción y reciclaje en el proteasoma. La presencia en cantidades elevadas de ubiquitina sugiere, entre otras cosas, la existencia de degeneración neuronal, modificaciones de los receptores de la superficie celular, canales iónicos y vías de secreción, etc.
En los últimos años, se ha sugerido que las inclusiones neuronales anormales de la proteína TDP-43 juegan un papel importante en la muerte neuronal en ELA, bien mediante un efecto tóxico intrínseco, o bien mediante el “secuestro” de la proteína TDP-43 y la evitación de sus funciones normales: actúa como proteína de unión al ARN en células cerebrales y participa de su correcto procesamiento; una de sus funciones claves es el reclutamiento de los ARNm transcritos en gránulos que se generan cuando la célula está sometida a estrés, lo cual facilita la supervivencia celular mediante la traducción de los ARNm de proteínas necesarias para combatir el estrés celular, mientras se silencian el resto de ARNm (Oskarsson et al., 2018). Sin embargo, aún permanece sin resolverse la cuestión de si la ELA es una proteinopatía, una ribonucleopatía o una mezcla de ambas; además, algunos de los mecanismos patológicos implicados parecen conectar de algún modo la ELA con la demencia frontotemporal y con otras patologías neurodegenerativas, como las atrofias cerebelares y la distrofia miotónica.
Por otra parte, entre los factores ambientales y de estilo de vida estudiados, no se ha establecido un vínculo irrefutable con la ELA para ninguno de ellos. Entre los analizados de forma de forma sistemática, destaca el tabaquismo, planteado como un factor de riesgo débil, especialmente en mujeres. Un análisis combinado (Wang et al., 2011) de los datos procedentes de 5 cohortes que incluyeron un total de 832 pacientes con ELA identificados de entre un conjunto de 562.804 varones y 556.276 mujeres, asociaba el tabaquismo con un incremento del riesgo relativo del 44% (RR= 1,44; IC95% 1,23 a 1,68; p< 0,001) de padecer ELA entre los exfumadores y del 42% para los fumadores aún en activo (RR= 1,42; IC95% 1,07 a 1,88; p= 0,02); asimismo, se observó que entre los fumadores de toda la vida, el riesgo era significativamente mayor (p= 0,03) cuanto más jóvenes se iniciaron en el tabaquismo. No obstante, hay cierta controversia al respecto, pues otros estudios posteriores de casos y controles no encontraron ninguna relación entre el tabaquismo y el riesgo de desarrollar ELA (Pamphlett et al., 2012).
También se ha considerado como una posible opción causal o desencadenante la exposición a químicos orgánicos, como pesticidas y otros solventes. Un meta‐análisis de 11 estudios que incluyeron a más de 2.000 pacientes fallecidos por ELA demostró que la exposición a los pesticidas incrementaba el riesgo de ELA en un 88% (RR= 1,88; IC95% 1,36 a 2,61), aunque no se especificó el tipo de pesticida en la mayoría de los estudios (Malek et al., 2012). Otros factores ambientales posiblemente asociados con ELA y/o su velocidad de progresión incluyen: la exposición previa a metales pesados (como cobre y mercurio), la exposición a campos electromagnéticos o shock eléctricos, antecedentes de lesión traumática (incluyendo traumatismo craneoencefálico), infecciones virales, un bajo índice de masa corporal o deficiencias en la dieta, nivel educativo bajo, ser un deportista profesional, participación en guerras o servicio militar4 o la exposición a N-metilamino-L-alanina (Wang et al., 2017).
—La ELA es la tercera enfermedad neurodegenerativa en incidencia, tras la demencia y la enfermedad de Parkinson—
Tal como se ha indicado, la ELA es la tercera enfermedad neurodegenerativa en incidencia, tras la demencia y la enfermedad de Parkinson, y la más frecuente de las enfermedades de motoneurona en el adulto. Globalmente, la incidencia oscila entre 1 a 2,5 casos por 100.000 habitantes y año (aunque en determinadas regiones, como ciertas islas del Pacífico Occidental, puede llegar a multiplicarse por 100), lo que supone que cada año se diagnostican unos 120.000 casos nuevos en todo el mundo. Tiene una incidencia ligeramente superior en hombres que en mujeres (ratio 1,5:1, diferencia que decrece conforme aumenta la edad) y afecta a personas de todas las razas y grupos étnicos, si bien las personas con más probabilidad de desarrollar la enfermedad son las de raza blanca y de etnias no hispanas.
La prevalencia se estima en torno a 5-6 casos por cada 100.000 habitantes, siendo relativamente uniforme en los países occidentales, con un total de aproximadamente medio millón de personas conviviendo con la enfermedad en el mundo (Talbott et al., 2016). Por ejemplo, en EE.UU. los Centros para el Control y la Prevención de Enfermedades estimaron que, en 2016, entre 14.000 y 15.000 personas tenían ELA en ese país. La edad media de inicio de la ELA se encuentra entre los 58-66 años, con un pico de incidencia a los 70-75 años y una disminución en edades superiores. Los casos de ELA con origen hereditario presentan un inicio a edades más tempranas (unos 10 años antes en muchas ocasiones).
En España, la incidencia es similar al resto de países desarrollados (en torno a 1,4 casos por cada 100.000 habitantes y año); sin embargo, nuestro país presenta una prevalencia de 8 casos por 100.000 habitantes, superior a la global, probablemente debido a la mayor esperanza de vida. Con estos datos, se estima que en España viven con ELA unas 4.000 personas (un 5% son formas familiares de la enfermedad) y cada año se diagnostican unos 900 casos de ELA, esto es, unos 3 casos diarios (Camacho et al., 2014; Fundación Luzón, 2017). La edad media de inicio es inferior a la observada a nivel mundial, ya que aparece fundamentalmente entre los 40 y 70 años, con una media situada entre los 48 y los 50 años. Por tanto, la mayoría de las personas recién diagnosticadas en España se encuentran en edad laboral.
Siguiendo los criterios definitorios en la UE, donde se calcula que está afectada el 0,05% de la población, la ELA se considera una enfermedad rara (menos de 1 caso por cada 2.000 personas o 50 casos por cada 100.000). Mientras que su incidencia es similar a la de otras enfermedades consideradas comunes, como la esclerosis múltiple (de origen autoinmune), la prevalencia de la ELA es mucho menor debida a la reducida esperanza de vida desde el momento de inicio de los síntomas: 6-8 casos/100.000 habitantes vs. 80-90 casos/100.000 habitantes en el caso de la esclerosis múltiple.
En este sentido, hay que recordar que la ELA es una patología incurable y de carácter fatal desde el inicio de la enfermedad. La esperanza de vida de ronda actualmente los 2-5 años (una media cercana a 3 años, variable según la publicación consultada) desde el inicio de los síntomas: el 50% fallece en menos de 3 años, un 80% en menos de 5 años, y la mayoría (más del 90%) en menos de 10 años. Así, menos del 20% de los pacientes supera los 5 años tras el diagnóstico (mayor probabilidad entre los casos diagnosticados en pacientes entre 20 y 40 años de edad), y solo un 10% de los pacientes llega a vivir ≥ 10 años después del diagnóstico. En España, la mortalidad anual ajustada por edad de la ELA se sitúa cerca de los 1,5 fallecimientos por cada 100.000 habitantes, cifra muy similar a la de nuevos diagnósticos y que ha ido aumentando paulatinamente desde el año 2.000 (probablemente relacionado con una mejor codificación y conocimiento de la enfermedad).
En vista de lo anterior, además del extraordinario impacto personal, la ELA es una patología que conlleva un alto impacto socioeconómico. Las crecientes necesidades de los pacientes obligan a un esfuerzo extraordinario a los cuidadores (generalmente, la propia familia) y al Sistema Nacional de Salud, habida cuenta de que la atención precoz contribuye decisivamente en los propios pacientes (pero también en sus cuidadores) para prevenir o paliar el deterioro y afrontar el obligado proceso de dependencia.
Como ocurre en otros cuadros neurodegenerativos –por ejemplo, las demencias–, el cuidador principal frecuentemente tiene que dedicar mucho tiempo al paciente, buscando el difícil equilibrio entre un trato afectuoso y la firmeza que requieren ciertas actitudes del paciente (autocompasión o rebeldía), comprensibles por su impotencia funcional. Cuando el cuidador principal es un familiar –lo más frecuente– es muy probable que tenga que reducir su jornada laboral o incluso abandonar su puesto de trabajo. Solo en casos excepcionales es posible o se plantea un cambio de domicilio debido a los problemas de movilidad asociados a la ELA, pero sí es frecuente que se realicen obras de adaptación del hogar para facilitar el desenvolvimiento del paciente y la del cuidador.
En ese contexto, la educación y el soporte económico, sanitario, social y emocional a la familia por parte de los servicios asistenciales públicos y las asociaciones de pacientes son fundamentales en el proceso de atención al paciente, de gran relevancia para combatir la posible incapacidad de la familia para responder adecuadamente a las demandas y necesidades del paciente. La cuestión económica se sitúa, pues, como una prioridad innegable. Un reciente estudio realizado en un total de 636 pacientes con ELA en Gran Bretaña (Moore et al., 2019) sugería que el coste medio por paciente en un periodo de 3 meses es de unos 2.120 euros (aproximadamente 700 euros mensuales), suponiendo las estancias hospitalarias, en aquellos pacientes que la requirieron, la mayor parte del gasto (hasta el 36% del coste total). Además, los estados de mayor gravedad se asociaban con los mayores costes: el estado más favorable presentaba un coste medio de unos 1.230 euros, mientras que los pacientes más graves requerían un gasto de unos 3.740 euros. En España, el Sistema Nacional de Salud ofrece una cobertura amplia frente a las necesidades que ocasiona la ELA, si bien son cuantiosos los gastos extraordinarios no cubiertos por las ayudas públicas (obras de readaptación del hogar, adquisición de mobiliario, baja laboral del paciente o de sus cuidadores, etc.) y en no pocas ocasiones las ayudas públicas son insuficientes o tardías.
Los primeros síntomas de la enfermedad son debilidad y pérdida de fuerza localizada en uno o más músculos inervados por el mismo grupo de neuronas, en alguna de las extremidades o en la región cefálica (bulbar), y por lo general sin causa aparente (a veces, después de un traumatismo). El paciente puede también notar sensación de rigidez y torpeza de movimientos, que pueden predominar en una de las mitades del cuerpo (forma hemipléjica), pero que raramente tienen una manifestación más generalizada –afectando a las cuatro extremidades– en su inicio.
Los signos y síntomas que con mayor frecuencia causan la consulta inicial de los pacientes son:
Solo en un pequeño porcentaje de casos (un 2%) la ELA comienza a manifestarse a través de síntomas respiratorios, asociados a debilidad muscular, especialmente del diafragma. La aparición de disnea desencadenada por esfuerzos relativamente pequeños (como subir las escaleras de casa) en una persona habituada a ellos, puede ser el primer síntoma de la enfermedad.
En definitiva, la característica clínica inicial más notable es la asociación en un mismo territorio muscular (en uno o más miotomas adyacentes) de síntomas y signos que reflejan la afectación de las neuronas motoras tanto superiores como inferiores (Figura 1). Progresivamente, puede desarrollarse una espasticidad en las extremidades atrofiadas y debilitadas, afectando a la destreza manual y a la marcha. Tras esto, la enfermedad tiende hacia una parálisis paulatina de toda la musculatura esquelética en un plazo aproximado de 2 a 5 años. En las fases avanzadas es cuando se afecta la musculatura respiratoria y el deterioro se vuelve más rápido; de hecho, los problemas respiratorios constituyen la principal causa de ingreso hospitalario y de mortalidad en los enfermos con ELA.
Ya que el proceso degenerativo deja indemnes a las restantes poblaciones neuronales, se conservan la funcionalidad del sistema sensorial, los mecanismos reguladores de control y de coordinación de los movimientos, y las funciones intelectuales (algunos pacientes pueden experimentar alteraciones neuropsiquiátricas, más ligadas a las limitaciones funcionales impuestas por la ELA que por su propia etiología). Las neuronas motoras relacionadas con los movimientos oculares y los músculos de los esfínteres tampoco suelen estar alteradas.
La debilidad muscular es el síntoma más relevante de la ELA, consecuencia de la muerte progresiva de motoneuronas. Comienza a percibirse cuando se ha perdido al menos un 50% de la población de neuronas motoras e inicialmente afecta frecuentemente solo a un grupo de músculos, para ir extendiéndose posteriormente ha-cia otros grupos a medida que progresa la enfermedad. La atrofia muscular, relacionada con la debilidad muscular, se debe a la degeneración de las fibras musculares por la denervación.
Otro síntoma típico son las fasciculaciones, contracciones espontáneas de un conjunto de fibras musculares inervadas por una misma motoneurona (unidad motora), debidas a alteraciones de la excitabilidad de la membrana de dicha neurona motora inferior o de su axón. Suelen ser observables a simple vista en brazos, pantorrillas, tórax o abdomen de los pacientes con ELA, especialmente si la capa cutánea de grasa no es abundante. A menudo el paciente no es consciente de ellas y se pueden observar con el músculo en reposo, siendo su frecuencia baja e irregular, repitiéndose a intervalos entre 1-5 s; se diferencian de las fasciculaciones benignas o fisiológicas, en que éstas últimas aparecen en personas sanas, son provocadas casi siempre por un sobreesfuerzo físico, tienen una frecuencia mayor (varias contracciones por segundo) y se repiten con cierta regularidad, desapareciendo al cabo de algunos minutos u horas).
Los calambres musculares (contracciones dolorosas involuntarias sostenidas de los músculos, de 30-45 s de duración) representan otro síntoma frecuente de la ELA, pudiendo afectar a cualquier músculo (cuello, mandíbula, manos, brazos, abdomen, muslos). Con el avance de la enfermedad, la reducción o pérdida de tono muscular (hi-potonía o atonía) y la arreflexia o ausencia de reflejos miotáticos (por los cuales, en condiciones normales, al estirar un músculo, éste responde con una contracción que se opone al estiramiento) son características de las parálisis periféricas; no obstante, en la ELA suelen mantenerse los reflejos en zonas con debilidad muscular, frecuentemente exaltados (respuestas exageradas ante estímulos pequeños) o patológicos.
La pérdida de neuronas motoras corticales produce torpeza y pérdida de destreza, y no debilidad muscular. Se suele manifestar como una sensación de agarrotamiento que dificulta los movimientos de las extremidades y que es debida a la pérdida del control inhibitorio que ejerce la vía corticoespinal sobre las neuronas motoras inferiores que inervan los músculos antagonistas. La ausencia de relajación de esos músculos conduce a un aumento del tono muscular, provocando espasticidad. Es característico el fenómeno de navaja, por el cual, al forzar la extensión de la articulación del codo o de la muñeca, se encuentra una resistencia anormal al inicio que luego cede bruscamente; en las extremidades superiores, predomina en los flexores y, en las inferiores, en los músculos extensores.
Los reflejos musculares profundos, regulados por el arco reflejo miotático a nivel medular, quedan liberados a consecuencia de la pérdida del control por parte de la neurona motora cortical, dando lugar a un estado de hiperreflexia; los reflejos patológicos, por los que un pequeño estímulo puede desencadenar varias contracciones repetidas y difundir incluso hacia otros grupos musculares próximos, son polisinápticos y su presencia corrobora la afectación de neurona motora superior. Entre los reflejos patológicos de las extremidades inferiores, el signo de Babinski (extensión dorsal del dedo gordo del pie, acompañada de la apertura en abanico de los demás dedos cuando se estimula mecánicamente la planta del pie) es el más conocido y, aunque solo se encuentra en un 50% de pacientes, su presencia es un signo inequívoco de ELA.
Mención aparte merecen los reflejos patológicos en los músculos de la vía aérea superior. En condiciones fisiológicas, la ventilación y la deglución se coordinan de manera que durante la deglución se produce una apnea que es seguida de una espiración para arrastrar hacia el exterior los posibles restos de alimentos no deglutidos. En algunos pacientes con ELA, este patrón está alterado y a la citada apnea puede seguirle una inspiración, lo que aumenta de forma significativa el riesgo de aspiración. Este reflejo debe ser tenido muy en cuenta ya que durante la ventilación mecánica no invasiva (VMNI) que se utiliza en fases avanzadas de la ELA, el paso del aire puede suponer un estímulo capaz de provocar contracciones de los músculos laríngeos, hasta el punto de que estas alteraciones pueden hacer fracasar la propia VMNI.
Por último, la labilidad emocional determina la presencia en el paciente de accesos de risa o de llanto de forma incontrolada, ante mínimos estímulos emocionales o incluso sin una causa aparente. Es un hallazgo frecuente en la ELA cuando hay afectación de la musculatura bulbar e implica la liberación de las vías corticobulbares por lesión de la neurona motora cortical (López-Casero et al., 2018).
El Ministerio de Sanidad, en su “Estrategia en Enfermedades Neurodegenerativas del Sistema Nacional de Salud” (MSSSI, 2016), reconoce cuatro formas clínicas de ELA según la semiología predominante:
Otros autores establecen hasta cinco fenotipos de ELA, cuyas principales características se resumen en la Tabla 1 (adaptada de Nowicka et al., 2019).
Además de lo anterior, todos los pacientes con ELA podrán presentar en su evolución una serie de cambios cognitivos de naturaleza variada, desde un leve déficit cognitivo –presente en hasta la mitad de los pacientes– hasta alteraciones neuropsicológicas más graves, como la demencia focal de tipo frontal (manifestada en < 5%). Son independientes del resto de la clínica neurológica típica de ELA, y más comunes –aunque no exclusivos– de fases iniciales de la enfermedad y de pacientes con síntomas bulbares. Se afectan fundamentalmente aquellas funciones neuropsicológicas reguladas mayoritariamente en el lóbulo prefrontal (corteza prefrontal dorsolateral y premotora).
Los trastornos neuropsicológicos más comunes en la ELA son: a) la disfunción ejecutiva, posiblemente la más frecuente y que determina algún grado de dificultad para formular objetivos y planificarlos, para la secuenciación, la autorregulación, la flexibilidad cognitiva y la resolución de problemas; b) los trastornos de la atención, que generalmente incluyen cierta reducción de la velocidad de procesamiento de la información, dificultad para atender a más de una cuestión a la vez, mantener la atención durante una actividad o centrarse en una tarea evitando las interferencias del entorno; y c) los problemas de memoria, que suelen aparecen en menor grado y consisten en la dificultad para aprender nueva información y/o para recordar lo ya aprendido.
El diagnóstico de la ELA es principalmente clínico. Un diagnóstico temprano es crucial para adaptar de forma precoz e individualizada el tratamiento y los cuidados que necesita un paciente. Sin embargo, los síntomas iniciales son muy inespecíficos, lo que da lugar, por un lado, a que el paciente suela tardar en consultar, y por otro, la baja incidencia de la patología hace que muchos profesionales no estén suficientemente familiarizados con la enfermedad y no la tengan en consideración en el diagnóstico (multitud de enfermedades más prevalentes producen síntomas similares a la ELA). Esa falta de información en los profesionales sanitarios a menudo induce a una aproximación diagnóstica errónea que da lugar a derivaciones incorrectas a otras especialidades médicas, pudiendo provocar tratamientos innecesarios e incluso contraproducentes para los pacientes. Casi ningún afectado de ELA es atendido directamente, en primera visita, por un neurólogo.
Al no existir una única prueba específica para diagnosticar la ELA, el neurólogo debe realizar un diagnóstico diferencial, que consiste en el estudio de la historia clínica, la realización de un examen clínico y una serie de pruebas que descarten otras enfermedades. Así, el tiempo medio que transcurre entre la aparición de los primeros síntomas hasta la confirmación del diagnóstico es de 9-14 meses. Algo más rápido puede ser en los casos de ELA familiar, a cuyo diagnóstico se llega cuando otros miembros de la familia padecen la enfermedad; se recomienda hacer un estudio genético en esos casos para dar un consejo genético a las familias. Cuando no hay antecedentes familiares, la aparición de un caso en la familia se entiende como esporádica, y en este sentido, los familiares del paciente no tienen un riesgo mayor que el resto de la población de padecer la enfermedad.
Los principales documentos de consenso en España (Barrera et al., 2017) recomiendan usar la versión validada en castellano de la escala revisada de valoración funcional de la ELA (Amyotrophic Lateral Sclerosis Functional Rating) (Salas et al., 2008) tanto en la valoración de la sintomatología para el diagnóstico como en la medición de la progresión en las sucesivas revisiones. Evalúa la discapacidad del paciente por áreas, se administra de forma rápida y se caracteriza por ser sencilla, facilitando la comprensión por pacientes y sus familiares o cuidadores; consta de 12 preguntas agrupadas en 4 dimensiones con relación a las actividades de la vida diaria: función motora gruesa, función motora fina, función bulbar y función respiratoria.
Los criterios diagnósticos de ELA vigentes (criterios de Arlie) añaden a los clínicos un apoyo diagnóstico basado en el estudio neurofisiológico del paciente, fundamentalmente mediante la realización de un electromiograma (técnica que registra la actividad eléctrica de las fibras musculares) para detectar la afectación de las neuronas motoras. También se utilizan pruebas complementarias de sangre u orina y/o resonancia magnética nuclear (RMN), estas últimas con el objetivo de descartar otras enfermedades (Lenglet et al., 2017). Por ejemplo, las RMN generalmente son normales en las personas con ELA, pero pueden revelar otros problemas que podrían estar causando los síntomas, como un tumor en la médula espinal, una hernia discal que comprime la médula, siringomielia o espondilosis cervical.
Igualmente, desde el inicio, el manejo adecuado de los problemas respiratorios requiere un diagnóstico de los mismos mediante una valoración clínica y funcional de los “problemas diana” en un servicio hospitalario especializado y capacitado: alteraciones de la musculatura bulbar, alteraciones de la tos y alteración de la ventilación.
Por el momento, no se dispone de un tratamiento curativo para la ELA, y dado su carácter progresivo e irreversible, el manejo terapéutico se centra en el control de los síntomas, en prevenir complicaciones innecesarias y en la mejora de la calidad de vida de pacientes (y cuidadores). El abordaje debe ser individualizado e integral y comenzar desde el momento mismo de la comunicación del diagnóstico al paciente y a sus familiares.
Para alcanzar resultados óptimos, en el cuidado del paciente deben participar equipos multidisciplinares de profesionales de la salud, como médicos (neurólogos, neumólogos y gastroenterólogos), farmacéuticos, fisioterapeutas, terapeutas ocupacionales y logopedas, nutricionistas, trabajadores sociales, psicólogos clínicos y enfermeros de atención domiciliaria y cuidados paliativos. El objetivo último debe ser mantener a los pacientes con la mayor movilidad, comodidad e independencia posibles y durante el mayor tiempo posible. También se pretende prevenir o paliar los efectos psicológicos demoledores que la enfermedad provoca sobre el estado de ánimo de los pacientes, quienes observarán con plena consciencia cómo se anula progresivamente su autonomía motora, su capacidad de comunicación oral, la deglución de alimentos y bebidas, e incluso la respiración, mientras mantienen mayoritariamente intactos los sentidos y el intelecto.
La falta de un tratamiento etiológico auténticamente eficaz frente a la ELA, como ocurre en otras patologías neurodegenerativas, como la enfermedad de Alzheimer, queda tristemente comprobado al considerar la gravedad de su pronóstico. Durante las últimas décadas, un amplio número de fármacos experimentales demostraron prometedores resultados preclínicos para retrasar la progresión de la enfermedad en modelos animales, pero fracasaron a la hora de su evaluación en humanos o están aún transitando por ensayos de fases 1 a 3.
El único fármaco disponible en España para el tratamiento de la ELA tras su autorización en 1997 por la Agencia Española de Medicamentos y Productos Sanitarios es el riluzol (Figura 2), el único que ha demostrado un efecto positivo –moderado– en la desaceleración del progreso de la enfermedad y en la prolongación de la vida (en torno a una media de 3 meses) o el tiempo hasta la instauración de ventilación mecánica. Sin embargo, no hay evidencias de que el riluzol tenga un efecto favorable sobre la función o los síntomas motores, la función pulmonar, las fasciculaciones o la fuerza muscular.
De administración oral, se encuentra comercializado en nuestro país en forma de comprimidos (Rilutek® –la primera marca aprobada por la EMA– y EFG) y en forma de suspensión oral (Teglutik®), especialmente indicada esta última en pacientes cuya capacidad de deglución está comprometida. La dispensación y uso de todos los medicamentos con riluzol se producen en el ámbito hospitalario. La dosis diaria recomendada para adultos o pacientes de edad avanzada es de 50 mg cada 12 horas; el empleo de dosis mayores no parece incrementar significativamente el beneficio terapéutico.
Si bien no se conoce exactamente el mecanismo de acción del fármaco, se ha propuesto que el riluzol actúa a nivel del sistema neurológico, principalmente y según la dosis, evitando la excesiva activación de las neuronas motoras por el efecto aumentado del glutamato (es un antagonista de los receptores de glutamato), provocando además la inactivación de los canales de Na+ dependientes de voltaje. Su acción centrada en la hiperactividad glutamatérgica en las neuronas motoras confirma el planteamiento de que ése es uno de los mecanismos de estrés que contribuyen a la apoptosis neuronal en la ELA, junto a la alteración de la homeostasis del Ca2+ en su interior. A bajas concentraciones (< 1-10 μM), los efectos más prominentes de riluzol a nivel neuronal son la bajada de frecuencias de disparo repetitivo y la supresión de corriente de Na+ persistente; otros efectos incluyen la reducción de la liberación de neurotransmisores (a concentración de 1-20 μM) y la inhibición de los canales de Ca2+ dependientes de voltaje (10-40 μM).
Habida cuenta de que la supervivencia neuronal y la producción de factores neurotróficos a menudo depende de las interacciones sinápticas dependientes de la actividad entre neuronas presinápticas y sus dianas postsinápticas (tanto de otras neuronas como del músculo), la investigación en mayor profundidad de los efectos del riluzol sobre el disparo neuronal, la regulación de la liberación de transmisores sinápticos y la producción de otros factores neurotróficos se plantea como una interesante vía de investigación.
La última revisión de la Cochrane al respecto de la evidencia disponible sobre riluzol (Miller et al., 2012) examinó los 4 ensayos clínicos controlados realizados, que incluyeron un total de 1.477 pacientes, de los cuales 974 habían recibido el fármaco. En la evaluación de la supervivencia sin necesidad de traqueotomía como variable de eficacia, los resultados agrupados revelan que el tratamiento con 100 mg/día de riluzol produjo beneficios significativos en un grupo homogéneo de pacientes en los dos primeros estudios, con una reducción del riesgo del 20% (tasa de riesgo o HR= 0,80; IC95% 0,64 a 0,99; p= 0,042) en comparación con placebo, y sin hallarse muestras de heterogeneidad (p= 0,33). Cuando se agregó el tercer ensayo (que incluyó pacientes de mayor edad y más gravemente afectados), el efecto global del tratamiento siguió siendo mostrando beneficio, aunque más reducido (HR= 0,84; IC95% 0,69 a 0,99; p= 0,046), pero se observaron pruebas de heterogeneidad. En términos clínicos, esto representa una mejora del 9% en la probabilidad de sobrevivir un año con el tratamiento con riluzol (58% vs. 49% en el grupo placebo) y un incremento de la supervivencia media de 3 meses (11,8 vs. 14,8 con placebo).
Se estima que el NNT (número de pacientes que deben ser tratados) para retrasar una muerte 12 meses es de 11, lo que sugiere una eficacia muy modesta. Además, tiene poco efecto cuando se administra en las fases más avanzadas de la enfermedad y no está claro durante cuánto tiempo debe administrarse el tratamiento. En cualquier caso, se asume que el tratamiento debe instaurarse tan pronto como se diagnostique la ELA.
En términos de seguridad, el riluzol no parece ser especialmente tóxico, siendo raros los efectos adversos graves. No obstante, con una frecuencia superior al 10% pueden aparecer: astenia o fatiga (26% vs. 13% con placebo), náuseas y elevación de los niveles de transaminasas hepáticas (que ocurre habitualmente a partir de los 3 meses de tratamiento, pero suele ser transitoria y disminuir paulatinamente sin necesidad de suspender el tratamiento en la mayoría de los casos). Con menor frecuencia (1-10%) pueden aparecer: diarrea, dolor abdominal, vómitos, cefalea, mareos, parestesia, somnolencia y dolor.
Por otro lado, también merece una mención aquí la edaravona (Radicava®), un fármaco de administración intravenosa aún no aprobado en Europa por la EMA, pero que fue autorizado para el tratamiento de la ELA en 2015 en Japón y en 2017 por la FDA en EE.UU. Previamente se había autorizado su uso en el mercado asiático para el tratamiento de las consecuencias a largo plazo derivadas del ictus.
Se trata de un fármaco antioxidante (Figura 3) que actúa disminuyendo el estrés oxidativo y que ha sido evaluado en un ensayo clínico en fase 2 y 3 estudios en fase 3. El estudio que condujo a su autorización fue el último de ellos, que incluyó a 137 pacientes con ELA de un total de 31 hospitales japoneses con edades comprendidas entre 21 y 75 años. Basado en los resultados de un ensayo previo, los criterios de inclusión en este estudio fueron muy estrictos, excluyéndose aquellos pacientes con formas extremas de la enfermedad, con alteración de la función respiratoria o con tiempos de evolución de más de 2 años. Los pacientes fueron tratados por vía intravenosa con edaravona 60 mg/día durante 6 ciclos. El análisis de los resultados evidenció que aquellos pacientes tratados con el fármaco presentaban un menor deterioro a nivel de la escala funcional ALSFRS-R (el cambio medio en la puntuación fue de -5,01 ± 0,64 puntos vs. -7,50 ± 0,66 puntos con placebo; p= 0,001) y mejores puntuaciones en el cuestionario de calidad de vida ALSAQ-40 en comparación con placebo.
No obstante, los ensayos con edaravona presentan ciertas limitaciones: a) se realizaron en población japonesa6 y con criterios de inclusión muy restrictivos, demostrando su eficacia solo en pacientes con corta evolución de enfermedad y menor gravedad de la misma; b) en el último de los estudios analizados, los resultados fueron siempre en contexto de uso concomitante de riluzol; y c) el efecto descrito sobre la progresión de la enfermedad en el estudio analizado es de solo 2 puntos sobre una escala funcional –la ALSFRS-R– de 48 puntos (eficacia muy modesta), a 6 meses de seguimiento y sin presentar datos sobre el seguimiento a largo plazo, por lo que no se aportan datos sobre el efecto en la supervivencia ni sobre otros biomarcadores.
Además, conviene subrayar que los estudios exploratorios previos no evidenciaron ningún efecto beneficioso en fases moderadas o avanzadas de la enfermedad o en pacientes con afectación de la función respiratoria. En relación al perfil toxicológico, la administración de edaravona no está exenta de efectos adversos; los más severos descritos son reacciones cutáneas (hematomas) y alteración de la marcha.
El manejo de los síntomas de la ELA se ha revelado como fundamental para preservar durante el mayor tiempo posible la funcionalidad cotidiana del paciente. Se suelen emplear fármacos que son prescritos por su indicación habitual, pues salvo excepciones, no hay estudios específicos para su uso en ELA. El manejo de los síntomas principales se resume a continuación (López-Casero et al., 2019).
SIALORREA
El exceso de salivación en los enfermos de ELA es debido al deficiente cierre de los labios, falta de control postural de la cabeza y pérdida de la capacidad de deglución, así como al cierre defectuoso del velo faríngeo y a alteraciones del sistema vegetativo. Este signo, que resulta muy molesto para los pacientes, puede facilitar el desarrollo de estomatitis o de infecciones fúngicas en la boca. Se suele manejar con fármacos anticolinérgicos y, sobre todo, con el antidepresivo tricíclico amitriptilina, si bien no se dispone de estudios específicos al respecto. Normalmente se inicia con dosis nocturnas bajas y se irá subiendo a 10-25 mg/8 h, aunque pueden requerirse dosis superiores (hasta 50 mg/8 h).
Una alternativa a la amitriptilina es el uso de un anticolinérgico puro, como la atropina en gotas al 0,5‐1% de administración sublingual (3-4 veces al día, debido a la corta duración de su acción), especialmente recomendado cuando aparece concomitantemente sequedad de boca. En España no se dispone de presentaciones comerciales adecuadas a esa concentración, pero pueden prepararse las soluciones mediante formulación magistral. Cuando estos tratamientos pierden eficacia o son insuficientes, se recomienda el uso de toxina botulínica mediante infiltración en las glándulas salivares: los resultados son, en general, satisfactorios, con una duración máxima de 4-6 meses y, en general, las inyecciones son bien toleradas.
Si la farmacoterapia se vuelve ineficaz, se recurre a la radioterapia de las glándulas salivares, que ha demostrado resultados muy satisfactorios. La cirugía no está indicada debido a efectos secundarios como la producción excesiva de moco denso.
DIFICULTAD EN LA EXPECTORACIÓNEn general, los pacientes con insuficiencia respiratoria o bulbar suelen presentar dificultades para llevar a cabo una limpieza adecuada del árbol respiratorio, o sea, falla la expectoración, lo cual provoca la acumulación de moco, siendo un factor pronóstico negativo sobre todo en pacientes con ELA sometidos a ventilación mecánica no invasiva. Aunque se ha sugerido el empleo de fármacos mucolíticos como la guaifenesina o la N-acetilcisteína (ésta en dosis de 200-400 mg/8 h), o broncodilatadores como el bromuro de ipratropio o la teofilina, lo cierto es que no se dispone de estudios clínicos específicos en pacientes con ELA y su eficacia parece limitada. Los mucolíticos solo se recomiendan cuando el paciente es capaz de toser adecuadamente. También se considera beneficioso el uso de dispositivos insufladores/exsufladores –particularmente en pacientes con ELA afectados por neumonía– y los humidificadores ambientales.
DOLORES Y CALAMBRESEn fases avanzadas de la enfermedad, un elevado porcentaje de los pacientes (entre 50-70%) puede experimentar dolor provocado por calambres, contracturas musculares, atrofia muscular o espasticidad que aparece generalmente antes de dormir, pero cuyas características, origen, localización e intensidad varían ampliamente. En su abordaje, se recomienda el escalado de la OMS, comenzando por AINES y analgésicos menores hasta concluir con los opiáceos.
Para minimizar el impacto de los calambres (también de las fasciculaciones, aunque éstas son muchas veces imperceptibles y mejor toleradas) se recomienda realizar estiramientos, así como masajes, mantener el músculo afectado caliente y estirarlo hasta que el dolor se alivie. Si se hace necesario introducir fármacos, y aunque no hay grandes evidencias que respalden su uso, la guía NICE recomienda el sulfato de quinina como primera elección; si no fuera efectivo o bien tolerado, se debería pasar a una segunda línea de baclofeno, o bien una tercera con tizanidina, gabapentina o dantroleno.
ESPASTICIDAD
Aunque la fisioterapia es, sin lugar a dudas, el mejor tratamiento para combatir la espasticidad que sufren los pacientes con ELA, comúnmente se recurre a otras intervenciones, tales como hidroterapia, calor, frío, ultrasonidos o electro-estimulación. Cuando éstas no son efectivas, suele recurrirse a los mismos fármacos comentados para los calambres, siendo de referencia el baclofeno; por lo general, se administra por vía oral a dosis crecientes (a partir de 5 mg/8 h), pero su eficacia parece mayor –en términos de control de dolor y de mejora de la calidad de vida– en administración intratecal. El tratamiento farmacológico deberá hacerse de forma progresiva, pudiéndose emplear, alternativamente a baclofeno, la tizanidina (6-24 mg/día), gabapentina (900-2.400 mg/día), memantina (10-60 mg/día), dantroleno (25-100 mg/día), tetrazepam (100-200 mg/día) o diazepam (10-30 mg/día); ninguno de ellos ha sido investigado específicamente en ELA.
LABILIDAD EMOCIONAL Y OTROS TRASTORNOS MENTALES
La presencia de episodios de risa y/o llanto incongruentes puede llegar a afectar a más de la mitad de los pacientes con ELA, incluso en ausencia de síntomas motores. Si fuera necesario tratar estos signos, hay datos clínicos que demuestran la eficacia del tratamiento con antidepresivos tricíclicos (amitriptilina en dosis de 50-75 mg/día) y con inhibidores de la recaptación de serotonina (citalopram, fluvoxamina). Pero, sobre todo, cabe destacar que, en base a dos ensayos clínicos, la FDA autorizó el uso de dextrometorfano/quinidina (20/10 mg), aunque no está comercializado en Europa; en España puede elaborarse como fórmula magistral en farmacia hospitalaria.
Por otra parte, dado el carácter progresivo y el pronóstico de la ELA, es previsible que en algún momento de la evolución aparezca sintomatología de depresión o ansiedad, no solo en pacientes sino también en los propios cuidadores o familiares. Para el tratamiento de la depresión se utilizará amitriptilina, los inhibidores de la recaptación de serotonina (ISRS: fluoxetina, paroxetina, escitalopram, citalopram) o mirtazapina a las dosis habituales, ajustando el fármaco y su pauta posológica de forma individualizada7 según el deterioro cognitivo, edad u otras patologías del paciente. En el caso de que el síntoma predominante sea la ansiedad, que suele ocurrir durante las fases de diagnóstico y en la fase terminal, se tratará con benzodiacepinas como lorazepam sublingual o diazepam.
INSOMNIO Y FATIGA
El insomnio –originado por diferentes causas (depresión, calambres/dolor, etc.)– suele ser frecuente (30-50%) en los pacientes con ELA, especialmente durante la última fase de la enfermedad, y puede generar un considerable malestar, empeorando el cansancio y la debilidad. Como tratamiento sintomático en pacientes con ELA, se han obtenido buenas respuestas con amitriptilina, mirtazapina o zolpidem. Asimismo, para el tratamiento de la fatiga se obtiene una respuesta favorable con el uso de modafilino.
ALTERACIONES DEL TRACTO GASTROINTESTINAL
La inmovilidad y el tratamiento farmacológico de otros síntomas de la ELA (con opiáceos, amitriptilina, etc.) son factores que pueden hacer que los pacientes sufran estreñimiento. Así, se recomiendan medidas dietéticas (aumentar la ingesta de líquidos y fibra) pero, en las fases más avanzadas, puede ser necesario el uso de laxantes estimulantes del peristaltismo (senósidos, bisacodilo), osmóticos (lactulosa, lactitol) o de acción local (supositorios de glicerina, enemas, etc.).
De igual modo, es posible la aparición de cuadros de reflujo gastroesofágico (ERGE) –por afectación del músculo diafragmático a nivel del esfínter esofágico inferior– que pueden provocar, además de las molestias asociadas, la aparición de disnea nocturna o dar lugar a una aspiración pulmonar del contenido gástrico, especialmente peligrosa. El tratamiento incluye medidas posturales, fármacos procinéticos (como metoclopramida) y antisecretores gástricos (omeprazol y similares).
TROMBOSIS VENOSA PROFUNDA
Los pacientes con ELA presentan mayor riesgo de TVP, relacionado con un mayor grado de inmovilidad y problemas respiratorios (independiente de la edad del paciente), con una incidencia anual que alcanza casi el 3%. Si bien de modo profiláctico se recomiendan medidas no farmacológicas, como la elevación de las piernas o el uso de medias compresivas, si aparece TVP se debe recurrir a la farmacoterapia, concretamente al uso de anticoagulantes orales; no hay estudios sobre la anticoagulación profiláctica en estos pacientes, por lo que no está recomendada.
Es importante tener en cuenta que, en tanto que es una enfermedad rara, los pacientes con ELA y sus cuidadores experimentan una sensación de aislamiento e incomprensión, que se une a la sensación de soledad y de exclusión social, cultural y económica, por el hecho de ser una enfermedad grave, degenerativa e invalidante. Como ya se ha sugerido, la pérdida del control de los movimientos voluntarios conduce a una progresiva dependencia de los pacientes por sus cuidadores, paralelo a la aparición de sentimientos de inutilidad, frustración y amargura por ser una carga para sus seres más queridos. Desgraciadamente, no todos los entornos familiares y sociales responden bien ante el enorme esfuerzo económico, físico y emocional que supone la ELA.
Las notables modificaciones en la vida del paciente con ELA, de su familia y entorno social requieren indispensablemente de una serie de cuidados generales en la asistencia sanitaria rehabilitadora, que incluirá disciplinas como la fisioterapia, logopedia, psicología o la terapia respiratoria, fundamentales para retrasar la progresión de la enfermedad y mejorar la calidad de vida. El abordaje será progresivo, adaptados a la evolución de la enfermedad y orientado a los objetivos concretos. La Guía para el manejo clínico de la ELA, publicada por la Federación Europea de Sociedades Neurológicas (Andersen et al., 2012) orienta hacia una serie de recomendaciones que se recogen a continuación.
Las complicaciones respiratorias, debidas a la afectación del diafragma (disnea y ortopnea), bulbar (infecciones por aspiración), o a la debilidad de los músculos espiratorios (menor efectividad de la tos, conducente a disnea e infecciones repetidas), representan la causa más frecuente de morbimortalidad en los pacientes con ELA. Resulta fundamental, por tanto, su diagnóstico precoz, que permita un tratamiento eficaz temprano, ya que la evolución de los procedimientos y ayudas para conseguir una tos efectiva y una ventilación alveolar adecuada han logrado mejorar la supervivencia y la calidad de vida de los pacientes, disminuyendo el número de hospitalizaciones. Los pacientes se ven muy beneficiados de la realización de ejercicios de fisioterapia respiratoria, recomendándose el uso de un aspirador portátil y de aerosoles en el domicilio a medida que la enfermedad avanza.
La ventilación mecánica no invasiva (VMNI) es uno de los procedimientos fundamentales en el manejo de los pacientes con ELA. Sin necesidad de acceder a la tráquea, mejora la ventilación alveolar y, con ello, los síntomas respiratorios, los trastornos del sueño y la supervivencia. En primera instancia, se recurre a la VMNI nocturna cuando el paciente es incapaz de dormir, si se produce hipoxemia nocturna excesiva o si los signos y síntomas de hipoventilación afectan significativamente al bienestar del paciente. En general, se recomienda a partir de una presión alveolar de CO2 > 45 mmHg acompañada de alteraciones clínicas o si supera los 50 mmHg con o sin manifestaciones clínicas. A medida que la debilidad muscular progresa, se prescribirá también de manera diurna para el alivio de la disnea.
La ventilación mecánica invasiva (VMI) –la traqueotomía– se utiliza cuando la VMNI no es eficaz o factible, y su importancia es tal que puede prolongar significativamente la supervivencia de los pacientes con ELA, en ocasiones durante varios años. Como contrapartida, tiene un elevado impacto emocional y social para pacientes y cuidadores, siendo la preocupación inicial la pérdida del habla (en algunos casos, es posible hablar cuando se tapa la cánula y el paciente está desconectado del respirador). Algunos autores apuntan a que más de la mitad (56%) de los pacientes con distrés respiratorio no reciben una adecuada asistencia respiratoria, un 73% no reciben fisioterapia respiratoria y únicamente uno de cada cinco (22%) pacientes tiene oxígeno en su domicilio; la traqueotomía se practica a un 8% de los pacientes con ELA (Mora et al., 2008).
El estado nutricional, cuantificado generalmente mediante la evolución del peso corporal, es uno de los principales factores de predicción de la evolución de la enfermedad y de supervivencia del paciente: una pérdida de peso > 5% en 1 mes, > 10% en 6 meses o un IMC < 19 serán indicativos de déficit nutricional. Los problemas relacionados con la pérdida de peso, la debilidad muscular resultante y las alteraciones nutricionales (que pueden incluso alterar la respuesta inmunitaria, con mayor propensión a sufrir infecciones) se relacionan fundamentalmente con la disfagia, aunque en ocasiones también pueden ocurrir en su ausencia. Debe hacerse una valoración nutricional periódica por un nutricionista, aún en ausencia aparente de síntomas; algunos estudios sugieren que el aporte de una dieta hipercalórica favorece a los pacientes.
Para paliar las consecuencias de la disfagia se recomienda adoptar, de forma precoz y progresivamente, medidas como: evitar alimentos que puedan provocar atragantamientos, fragmentar las comidas, cambiar la consistencia de los alimentos (por ejemplo, dieta triturada) o emplear alimentos de fácil masticación y deglución. La nutrición enteral mediante gastrostomía percutánea se considerará si las medidas anteriores se vuelven ineficaces, sin que ello suponga el abandono total de la ingesta por vía oral, y antes de que los problemas de malnutrición o insuficiencia respiratoria incrementen el riesgo del procedimiento; será de vital importancia realizar desinfecciones diarias (con clorhexidina) del estroma y de la sonda, que deberán sustituirse periódicamente en función del deterioro que presenten. Se estima que más del 70% de pacientes con disfagia no reciben asistencia o información nutricional y el 60% de pacientes con disfagia grave no tiene gastrostomía.
Es un pilar clave en el tratamiento individualizado del paciente con ELA, que debe incluirse en su plan de cuidados desde el diagnóstico para corregir las posibles alteraciones de la postura, prevenir el dolor y disminuir la rigidez muscular; permite mantener la independencia funcional, especialmente para prevenir las caídas, y facilitar la marcha mediante determinadas ayudas técnicas. El ejercicio físico debe incluir inicialmente ejercicios como natación o caminar, para trabajar posteriormente –cuando estas actividades ya no se puedan realizar– un programa de ejercicios de movimiento de articulaciones, reducción de la rigidez y estiramientos. Es importante dotar a los cuidadores de formación específica para realizar los movimientos a los pacientes, sobre todo aquellos para aliviar síntomas como los calambres y la espasticidad.
La colaboración de la fisioterapia es esencial, dado que los pacientes con ELA suelen presentar una baja tolerancia al ejercicio físico, fatigándose rápidamente. Por ello, estos ejercicios deben tener una breve duración (30-45 minutos), pero pueden repetirse varias veces a lo largo del día (normalmente se dividen en 2 o 3 sesiones diarias). Aun siendo de vital importancia, casi la mitad de los pacientes no reciben ninguna forma de fisioterapia y los que la reciben es por un tiempo limitado, mayoritariamente menos de 3 meses (Mora et al., 2008).
La intervención de un logopeda, como especialista del habla, la deglución y la comunicación, busca potenciar los músculos orofaciales, de articulación y respiratorios para favorecer el entendimiento y mejorar la capacidad de comunicación, pues la mayoría de los pacientes –sobre todo con ELA de tipo bulbar– presentan hipofonía o disartria (ésta última es una de las limitaciones que más frustración genera), que se empiezan a manifestar con el uso de frases cortas y lentas, pausas inapropiadas, voz más grave, menor volumen, etc. Los ejercicios logopédicos también deben ser regulares y moderados, a fin de evitar la fatiga, y preferiblemente implicarán al cuidador directo, para que emplee estrategias en la conversación con el paciente, que se han revelado eficaces, tales como: confirmar las preguntas y las respuestas dadas, ofrecer pistas del tema sobre el que quiere expresarse, mantener una actitud de humor ante los malentendidos que pudieran generarse, etc.
Para facilitar la comunicación se dispone de un amplio abanico de ayudas técnicas, que van desde una simple pizarra plástica hasta amplificadores de voz, para los casos de hipofonía severa, avisadores acústicos, o incluso dispositivos con tecnología avanzada activados con la mirada o con pulsador. Pero se estima que 3 de cada 4 pacientes no recibe logopedia (la mayoría –73%– de los que sí la recibían, lo hacían en centros privados) y, entre los pacientes con disartria severa, esa misma proporción no disponían de comunicador informático, electrónico o manual.
Cerca de la mitad de los pacientes con ELA pueden experimentar algún tipo de alteración cognitiva y/o del comportamiento, que debuta incluso antes que los síntomas motores, lo que tiende a empeorar el pronóstico. Además, los pacientes con ELA y sus cuidadores presentan altos niveles de estrés y otra sintomatología afectiva (ansiedad, depresión, etc.) asociados a la enfermedad. En general, pacientes y familias precisan, sobre todo al inicio de la enfermedad, apoyo psicológico y soporte emocional, orientación y apoyo, con el fin de reducir los estados de ánimo adversos y favorecer la adaptación personal y social. Sin embargo, se calcula que solo 1 de cada 4 recibe psicoterapia profesional sistemática.
Por último, la terapia ocupacional juega también una función importante en la instrucción del paciente y sus cuidadores en lo relativo a facilitar el cumplimiento adecuado de las actividades cotidianas con la máxima autonomía posible. Frente a la pérdida de habilidad, en especial para manejar pequeños objetos (sostener un lápiz para escribir, coger los cubiertos para comer, asir un picaporte para abrir una puerta, etc.), existe un amplio catálogo de dispositivos y ayudas técnicas. Por ejemplo, cuando el deterioro motor avanza, conviene adaptar la ropa y el calzado con cierres fáciles (por ejemplo, con velcro), usar cinturones elásticos y otros dispositivos similares para facilitar los actos de vestirse y desvestirse. Los terapeutas ocupacionales también pueden sugerir dispositivos como rampas, aparato ortopédico, caminadores y sillas de ruedas que ayuden a las personas a conservar la energía y permanecer móviles.
En primer lugar, se debe subrayar que la investigación en cualquier enfermedad neurodegenerativa y de baja frecuencia posee sus peculiaridades, algunas de las cuales hacen que sea complejo su abordaje. Si bien la ELA comparte características –en cuanto a las vías moleculares de degeneración de las neuronas motoras– con otras enfermedades más “comunes” (como Alzheimer y Parkinson), su baja prevalencia limita el interés social que pueda atraer (relacionado con el potencial económico de los resultados de la investigación) y el acceso a un número de muestras suficiente para el desarrollo de estudios clínicos o traslacionales, tanto a nivel nacional como internacional. Ello dificulta la puesta en marcha de proyectos de investigación con suficiente potencial o solidez científica en comparación con enfermedades más comunes.
En cualquier caso, afortunadamente, existe una investigación activa de la ELA, sus causas y posibles tratamientos, en la que los pacientes participan activamente a través de ensayos clínicos. Multitud de grupos de investigación en los países desarrollados, así como en países emergentes como China o India, están dedicando sus esfuerzos a desgranar la problemática de esta patología. Además, se ha desarrollado una extensa colaboración internacional respecto al desarrollo de fármacos (los ensayos clínicos deben, inevitablemente, ser multicéntricos e internacionales) y en cuanto al hallazgo de las causas de la enfermedad, como el proyecto MinE (https://www.projectmine.com/es/) en el que se intentan hallar causas genéticas relacionadas con la ELA esporádica secuenciando el genoma completo de 15.000 pacientes y 7.500 controles procedentes de 17 países situados en 4 continentes (incluido España, con la participación coordinada de varios hospitales a través del Hospital Carlos III de Madrid).
En los últimos años se han investigado más de 60 moléculas como posible farmacoterapia para la ELA; pero, a pesar de los esfuerzos llevados a cabo, la mayoría han resultado clínicamente ineficaces (Petrov et al., 2017) y no hay aval científico que justifique la utilidad de los siguientes fármacos o sustancias: vitamina E, testosterona, antioxidantes como la coenzima Q10 o el Ginkgo biloba, inmunoglobulinas, ciclosporina, interferones, glatirámero, factores neurotróficos (como el factor neurotrófico derivado del cerebro o BDNF), ceftriaxona, gabapentina, minociclina, sales de litio, topiramato, oxandrolona, pentoxifilina, celecoxib, indinavir, etc. Recientemente se ha publicado un interesante artículo, de recomendable lectura, que revisa toda la investigación clínica desarrollada hasta ahora en torno a la ELA (Wobst et al., 2020).
A fecha de hoy, dos de las opciones evaluadas (masitinib oral y edaravona) parecen las más cercanas a confirmar su beneficio para pacientes con ELA. Habiéndose ya comentado la situación de edaravona intravenosa (autorizada fuera de Europa), se debe destacar que en estos momentos se encuentra en marcha la investigación clínica en población europea con una formulación oral de dicho fármaco (Treeway TW001), la cual ha completado con éxito un estudio de fase 1, pero de la que aún no se dispone de resultados confirmatorios de seguridad y eficacia.
Por otra parte, aún en línea de investigación y no autorizado en ningún país8, el masitinib es un inhibidor de tirosina cinasas (Figura 4) administrado por vía oral que se dirige a los mastocitos y macrófagos, células importantes para la inmunidad, a través de la inhibición de un número limitado de cinasas. El mecanismo de acción propuesto del masitinib en la ELA se basa en la inhibición de la proteína CSF1R, proporcionando un efecto neuroprotector y ralentizando la neurodegeneración.
Se realizó un estudio de fase 2 internacional, multicéntrico, doble ciego y con placebo en el que 382 pacientes que se encontraban en tratamiento con riluzol fueron asignados de manera aleatoria a recibir masitinib en dosis de 3 mg/kg/día, 4,5 mg/kg/día o placebo durante 48 semanas; el ensayo reclutó a una población de pacientes más amplia (menos restrictiva) y representativa en comparación con el estudio en que edaravona demostró beneficios. El objetivo principal era conseguir un descenso en la progresión de la escala funcional ALSFRS-R. Los datos analizados han indicado que los pacientes que recibieron la dosis de 4,5 mg tuvieron una caída menor en la escala funcional en comparación con placebo, además de una menor caída en otros objetivos como las escalas de calidad de vida y de función respiratoria. Sin embargo, no hubo diferencias significativas entre el grupo tratado con 3 mg/kg/día y el placebo (Fundación Luzón, 2017).
Recientemente se han divulgado resultados (Mora et al., 2020) para los análisis de los datos de este mismo ensayo tras la transición cegada a una fase 2/3, con un diseño prospectivamente definido en dos niveles basados en la progresión en la puntuación de ALSFRS-R desde el inicio de la enfermedad al estado basal. Tal abordaje permitió seleccionar una cohorte primaria de eficacia más homogénea (N= 99): aquellos pacientes que, tratados con la dosis más alta de masitinib, progresan de forma normal (cambio menor de 1,1 puntos/mes en la escala ALSFRS-R). En ese subgrupo, masitinib demostró un beneficio significativo sobre placebo (N= 102), con una diferencia en la variación de ALSFRS-R de 3,4 puntos (IC95% 0,65-6,13; p= 0,016), lo que se corresponde con una reducción de un 27% en la tasa de deterioro funcional. Todos los análisis de sensibilidad fueron consistentes y las variables secundarias (ALSAQ-40, capacidad vital forzada y tiempo hasta evento) respaldaron la superioridad de masitinib sobre placebo. Sin embargo, no se hallaron diferencias notables en comparación con la cohorte más amplia de pacientes con progresión normal y rápida tratados con la dosis alta, ni tampoco con la cohorte de pacientes tratados con la dosis baja (3 mg/kg/día). La tasa de eventos adversos emergentes durante el tratamiento fue del 88% con masitinib 4,5 mg/kg/día, 85% con 3,0 mg/kg/día y 79% con placebo, apareciendo eventos adversos graves en el 31, 23 y 18% de pacientes, respectivamente. Ninguna muerte se asoció al fármaco.
—El riluzol es, por ahora, el único fármaco con eficacia (modesta) contrastada para prolongar la vida
de pacientes con ELA—
Ha sido, pues, ése el primer ensayo clínico en mucho que tiempo (unas dos décadas) que arroja resultados positivos en ELA, aunque está pendiente el desarrollo de un ensayo confirmatorio de fase 3 con masitinib. Se ha especulado que los fracasos en la investigación clínica se deben, al menos en parte, a las diferencias en los mecanismos patofisiológicos subyacentes entre los pacientes con ELA. Así, se espera que una mejor comprensión de las bases moleculares y el desarrollo de modelos preclínicos más representativos facilite el hallazgo futuro de tratamientos eficaces.
Por el momento, las únicas causas conocidas y demostradas de la enfermedad son las de origen genético. Existen un número creciente de genes relacionados con la ELA gracias al estudio de familias en que aparecen, o han aparecido, varios casos con la enfermedad en familiares relacionados en primer o en segundo grado. El estudio de las causas genéticas permitirá comprender mejor los mecanismos de neurodegeneración. Así, debido a que los casos de ELA familiar son indistinguibles desde el punto de vista clínico de los casos mayoritarios de ELA esporádica, podemos utilizar el conocimiento de las causas genéticas en el resto de los pacientes, tanto para comprender el progreso, como para contrarrestar las causas con fármacos potencialmente terapéuticos. A modo de recordatorio, los genes más comúnmente mutados en pacientes con ELA son C9orf72, SOD1, TARDBP y FUS, y los mecanismos patogénicos propuestos incluyen la afectación del metabolismo del ARN y de las proteínas.
Además, hay que subrayar que el uso de modelos animales en la investigación de potenciales tratamientos se ve limitado por el hecho de que no se dispone de ningún modelo que en sí mismo mimetice perfectamente el perfil clínico y patológico de los pacientes de ELA; los estudios continuados de modelos más relevantes han provisto y proveerán de una mejor comprensión de la patogénesis de la enfermedad. El modelo de ratón más extendido y utilizado en todo el mundo es el modelo transgénico que sobreexpresa el gen humano SOD1 mutado, ratones que pueden ser rápidos, con mayor número de copias del gen humano (el modelo más usado) o lentos (menos utilizado, por necesitar más tiempo de mantenimiento en el estabulario), con la mitad de copias del gen humano mutado. Muchos de los comportamientos y cambios motores que experimentan recuerdan holgadamente a la ELA, pero se observan también en la mayoría de los modelos murinos de neurodegeneración.
El mayor problema es que, de todos los estudios preclínicos con terapias potenciales que han mostrado beneficio ralentizando el progreso de la enfermedad en el ratón, ninguno de ellos ha sido positivo al trasladarse a la realización de ensayos clínicos en pacientes con ELA. Debido a la estructura cerebral menos desarrollada que en primates, el ratón tiene grandes limitaciones en la expresión del deterioro neuronal en comparación con los complejos fenotipos de enfermedades neurodegenerativas en el ser humano. Además, es evidente la dificultad de transferir las dosis y tiempos de intervención, puesto que no es posible tratar a los pacientes antes de que los síntomas florezcan, generando bastante confusión en el desarrollo de los ensayos clínicos. A pesar de todo ello, los citados modelos de ratón transgénico con mutación de SOD1 siguen siendo esenciales en la investigación de las vías moleculares implicadas en la ELA y han asentado las bases del conocimiento de la patofisiología subyacente.
Una de las vías de investigación en boga es el mecanismo patogénico de excitotoxicidad mediada por glutamato, el principal aminoácido excitatorio (sintetizado en cerebro y médula espinal, fundamentalmente en astrocitos, pero también en neuronas y oligodendrocitos) que actúa como neurotransmisor9 en el SNC y es indispensable para las funciones cerebrales normales (movilidad, cognición, memoria y aprendizaje). La excitotoxicidad inducida por glutamato y el daño que provoca sobre las neuronas motoras puede deberse a niveles aumentados del glutamato sináptico y la excesiva estimulación de sus receptores o al aumento de la sensibilidad de las neuronas postsinápticas ante la acción del glutamato, que podría deberse a modificaciones en la homeostasis energética neuronal o a una mayor expresión de los receptores. Sobre esta vía actúa el único tratamiento autorizado para tratar la ELA en Europa, el riluzol, que interrumpe la transmisión glutamaérgica y atenúa la concentración de glutamato al antagonizar los receptores NMDA o AMPA y proteger de la excitotoxicidad.
La atención se desvió hacia el estrés oxidativo (y la respuesta celular al mismo), como otra de las vías estudio, precisamente cuando se descubrió el primer mecanismo subyacente de la neurodegeneración en la ELA: las mutaciones en el gen que codifica por el enzima citosólico SOD1. La investigación ha ido avanzando y sugiere que el estrés oxidativo en la ELA interacciona con –y también desequilibra– otros procesos patofisiológicos que contribuyen al daño de la neurona motora, incluyendo la excitotoxicidad, la alteración de la función mitocondrial, la agregación proteica, el estrés en el retículo endoplásmico y los cambios en la señalización celular en astrocitos y microglía. El análisis del líquido cefalorraquídeo y de tejidos post mortem del SNC de pacientes con ELA esporádica y ELA familiar ligada a SOD1 ha evidenciado la presencia más intensa –en comparación con sujetos sin ELA– de cambios bioquímicos resultado del daño de radicales libres (en proteínas, lípidos o ADN) o un metabolismo anormal de los mismos.
En este sentido, se han probado varios tipos de terapias antioxidantes, como la vitamina C, la N-acetil-L-cisteína (NAC), la porfirina de manganeso o la selegilina. Han probado su beneficio en modelos de ratón de ELA, fallando, sin embargo, en ensayos clínicos: los ensayos con antioxidantes se han realizado bajo condiciones subóptimas de diseño y calidad metodológica y, hasta la fecha, no son prometedores.
Otro mecanismo en investigación, estrechamente ligado con el anterior, es la disfunción de las mitocondrias, los orgánulos productores de energía de la célula que regulan vías celulares tales como la apoptosis, la homeostasis del calcio intracelular y la generación intracelular de radicales libres. Una amplia evidencia demuestra el deterioro de la función mitocondrial en la ELA, lo cual se pone de manifiesto en la elevada aparición de mutaciones en el ADN mitocondrial en corteza motora de pacientes con ELA esporádica, y en la disminución de dicho ADN en músculo y médula espinal. La agregación de la SOD1 mutante en la mitocondria parece ser una característica común en la ELA y su presencia puede alterar el equilibrio redox, provocar deficiencias de los complejos respiratorios mitocondriales y alterar la asociación del citocromo c con la membrana interna mitocondrial.
El enlace entre los defectos mitocondriales debidos a la función tóxica de la SOD1 mutante y la patogénesis de la ELA ha conducido a la búsqueda de terapias neuroprotectoras dirigidas específicamente a la mitocondria. Fármacos como olexosima, dexpramipexol o mitoQ han tenido buenos resultados en modelos animales, pero no han llegado a aportar buenos resultados en fase 3 para su aprobación por las agencias reguladoras. El dexpramipexol, un estereoisómero del pramipexol (agonista dopaminérgico utilizado en la enfermedad de Parkinson), llegó incluso a mostrar resultados positivos en un ensayo de fase 2, que sugerían un potencial terapéutico interesante; por ello, se llevó a cabo un amplio ensayo clínico de fase 3, doblemente ciego y controlado con placebo, que incluyó a 943 personas en 81 localizaciones de 11 países. Desgraciadamente, el análisis de los datos demostró que dexpramipexol no alcanzaba el objetivo primario de mejorar la valoración combinada de supervivencia y funcionalidad a los 12 meses de tratamiento (Cudkowicz et al., 2013).
A camino entre la lucha contra el estrés oxidativo y la disfunción mitocondrial, se evaluó el papel de la creatina, atendiendo a su efecto en la producción de ATP. Obtuvo buenos resultados en modelos murinos de ELA, con un incremento de la supervivencia. Sin embargo, varios ensayos clínicos realizados en pacientes dieron lugar a resultados contradictorios y poco claros. Una revisión sistemática de tres ensayos aleatorizados comparando creatina (5‐10 g/día) con placebo evidenció que los resultados agrupados no sustentaban ninguna diferencia en la supervivencia de los pacientes ni en las puntuaciones de la escala ALSFRS-R o en la capacidad pulmonar (Pastula et al., 2012).
Un último mecanismo patogénico propuesto se basa en la neuroinflamación, pues la muerte de las neuronas motoras en el cerebro y la médula espinal en la ELA se acompaña de una respuesta antiinflamatoria caracterizada por la activación microglial en las regiones afectadas del SNC. Por ello, se ha planteado el uso de terapias antiinflamatorias en el tratamiento de la enfermedad. La minociclina, una tetraciclina de segunda generación que posee propiedades antiinflamatorias, mostró un retraso en la neurodegeneración y un aumento de la supervivencia en diferentes modelos animales de ELA, como el de SOD1 mutante, en el cual redujo los niveles de apoptosis de las neuronas motoras mediante la disminución en la liberación de citocromo c y en la activación y proliferación microglial. Posteriormente, se realizó un ensayo aleatorizado de fase 3 (Gordon et al., 2007) en que se aleatorizaron 412 pacientes a recibir placebo o minociclina en dosis de 400 mg/día durante 9 meses. En comparación con placebo, los pacientes tratados con minociclina tuvieron un deterioro más rápido en la puntuación de la escala funcional ALSFRS-R, y también una tendencia –estadísticamente no significativa– hacia la caída de la capacidad pulmonar y de las pruebas musculares manuales con el tiempo; mostraron incluso un riesgo de mortalidad incrementado (hasta en un 30%) y una mayor incidencia de eventos adversos neurológicos y gastrointestinales.
Otro ejemplo de investigación en torno a la modulación de la neuroinflamación fue el estudio de fase 2/3 con celecoxib (Cudkowicz et al., 2006), un inhibidor de la ciclooxigenasa 2. Los resultados demostraron que celecoxib no detenía el declive de la fuerza muscular, no afectó la capacidad vital, tampoco influyó en la estimación del número de unidades motoras, ni en la escala ALSFRS-R o la supervivencia; se descartó, por tanto, un efecto beneficioso en los pacientes con ELA.
Entre los factores neurotróficos, destaca la utilidad del factor de crecimiento similar a insulina de tipo 1 (IGF-1), o su versión recombinante, la mecasermina, que han sido estudiadas en tres ensayos clínicos, cuyos resultados han sido agrupados en un meta‐análisis (Beauverd, 2012), incluyendo un total de 779 pacientes estudiados. En el primero de los ensayos (llevado a cabo en Europa sobre 183 pacientes), la variación media en la puntuación de la escala AALSRS (Appel Amyotrophic Lateral Sclerosis Rating Scale) después de 9 meses no fue estadísticamente significativa (‐3,3 puntos; IC95% ‐8,68 a 2,08); por otro lado, un estudio norteamericano (266 pacientes) mostró una diferencia significativa de ‐6,0 puntos (IC95% ‐10,99 a ‐1,01) y, al combinar ambos, la diferencia alcanzó significación estadística (‐4,75 puntos; IC95% ‐8,41 a ‐1,09). Sin embargo, en el tercero de los estudios, también realizado en Norteamérica (330 pacientes) pero con criterios más estrictos, no se encontraron diferencias estadísticamente significativas frente al placebo. En cualquier caso, no parece que se registrase un incremento de la supervivencia de los pacientes con ELA tratados con IFG‐1.
Entre las vías de investigación más modernas, destacan los estudios sobre células madre pluripotenciales. El desarrollo de modelos celulares ha arrojado luz sobre las vías moleculares asociadas con la ELA, proveyendo de alternativas a los modelos de roedores para los cribados farmacológicos de alto rendimiento. En fases preclínicas, se ha estudiado el trasplante de diferentes tipos de células en modelos de ratón, tales como las células madre neuronales (NSCs) y las células madre mesenquimales (MSCs)10. Por otro lado, las células madre inducidas pluripotentes (iPSCs), que poseen propiedades únicas como la autorrenovación y la diferenciación en múltiples subtipos celulares neuronales (neuronas, neuronas motoras, astrocitos y oligodendrocitos, entre otras), se pueden aprovechar como modelo de enfermedad para el hallazgo de fármacos y la realización de terapias de reemplazamiento autólogo (las células de la propia persona, diferenciadas y trasplantadas en algún lugar del SNC). De hecho, las neuronas motoras generadas de las iPSCs derivadas de fibroblastos de piel de pacientes con las formas esporádica o familiar de ELA suponen una técnica innovadora como modelo de enfermedad en la actualidad.
Los ensayos sobre el trasplante de células madre a dosis creciente en pacientes con ELA se encuentran aún en fases 1 y 2 de investigación clínica, habiéndose comprobado previamente que el trasplante de células madre de médula ósea en la médula espinal mejora la función motora de los animales y reduce la muerte de neuronas motoras, gracias a un efecto neurotrófico de las células madre. Un estudio de fase 2 (Glass et al., 2016) incluyó 15 pacientes divididos en 5 grupos de tratamiento que recibieron dosis crecientes de células madre, aumentando en cada nivel de dosis el número de células por inyección y el número de inyecciones; todos los participantes recibieron inyecciones bilaterales en la médula espinal cervical (C3-C5) y únicamente el último grupo recibió, adicionalmente, inyecciones en el cordón lumbar (L2-L4) a través de 2 procedimientos quirúrgicos separados. El objetivo principal fue evaluar la seguridad del tratamiento en estos pacientes, no demostrar la eficacia del mismo. Los efectos adversos se relacionaron con dolor asociado a la cirugía y con efectos secundarios de los medicamentos inmunosupresores, pudiendo concluir que los trasplantes se pueden realizar de manera segura incluso a altas dosis, sin que aceleren la progresión de la ELA. Aún es necesario desarrollar estudios específicos que prueben la posible eficacia clínica del trasplante de células madre, pero es una vía de investigación prometedora.
En el campo de las terapias avanzadas, indudablemente la terapia génica emerge como otra opción de casi obligado estudio, por la estrecha relación de algunos genes con la patogenia de la enfermedad. Por el momento, solo se dispone de ensayos en fase preclínica y con modelos de experimentación animal, en los que se han estudiado la utilización de genes que expresan factores neurotróficos humanos (utilizando agentes transfectores virales) o de la SOD‐1, con resultados alentadores. Asimismo, la técnica del silenciamiento génico o del RNA de interferencia (RNAi) se ha aplicado en modelos experimentales de la enfermedad y es una aproximación prometedora de un posible tratamiento, en principio para las formas familiares ligadas a mutaciones de la SOD‐1, que podría extenderse a otras alteraciones genéticas conocidas.
Todavía es pronto para saber la utilidad real de estas últimas técnicas en pacientes con ELA y los expertos creen que aún se está lejos de alcanzar la curación de la enfermedad, aunque cada vez más cerca de frenarla. En este sentido, actualmente están en curso diversos ensayos clínicos en la Unión Europea (EU Clinical Trials Register), algunos en España, en torno al tratamiento de la ELA. Se están barajando varias opciones en fase ≥ 2 del desarrollo clínico, como la colchicina, el ácido alfa-lipoico, el ácido taurodesoxicólico, la memantina (un antagonista de receptores NMDA), sirolimus, interleucina 2 a dosis bajas, eritropoyetina, tirasemtiv (activador de la troponina muscular), guanabenz (agonista adrenérgico α2 de acción central), filgrastim (versión recombinante del factor estimulante de colonias de granulocitos), arimoclomol (activador de chaperonas moleculares que estimula el normal reciclaje de proteínas) o levosimendan (inótropo que potencia la sensibilidad al calcio de las proteínas contráctiles), entre otras. También se está investigando un extracto de Cannabis sativa, el empleo de células madre multipotentes de origen osteomuscular y mesenquimatoso o la infusión intramuscular de células madre autólogas de médula ósea. Por último, este mismo año se ha iniciado un estudio en España con ravulizumab (un inhibidor de la activación del complemento). Por tanto, se espera contar con nuevos resultados en los próximos meses y años.
Entre las novedades de la investigación biomédica en torno a la ELA divulgadas en el último año, que recopila la Fundación Luzón en su sitio web11, podemos destacar las siguientes:
Por otro lado, a finales del año pasado, se publicaron resultados provisionales de un ensayo clínico de fase 1/2 –exploratorio de eficacia– con tofersen, un oligonucleótido antisentido que actúa a nivel genético bloqueando la síntesis de SOD1 y se plantea como terapia potencial para ralentizar la enfermedad en pacientes con ELA familiar con mutaciones en el gen SOD1. El análisis provisional de los datos mostraba una reducción significativa de los niveles de SOD1 en el fluido cerebroespinal en pacientes tratados con el fármaco, asociado a un cambio medio en la escala ALSFRS-R de -1,1 puntos hasta el día 85 de seguimiento (vs. -5,3 puntos en el grupo placebo). Actualmente se está realizando el ensayo en fase 3 de este tratamiento en 26 hospitales de Estados Unidos y Europa, y se espera que pueda completar el reclutamiento en el 2020.
—Una mejor comprensión de las bases moleculares de la enfermedad contribuirá al potencial hallazgo de tratamientos eficaces—
Entre las terapias avanzadas, cabe destacar que se ha iniciado un ensayo clínico de fase 1/2 de un nuevo tratamiento con células madre (llamado AstroRx) un tanto especial: no está orientado a generar nuevas motoneuronas sino a generar nuevos astrocitos. Los resultados parciales, solo basado en 5 pacientes, sugieren que la dosis más baja, sin efectos tóxicos registrados, parece reducir el ritmo de cambio en la escala ALSFRS-R en los 3 primeros meses, lo que supondría una progresión más lenta de la enfermedad. Se espera que los resultados de los demás grupos de pacientes en este ensayo estén listos en la primera mitad de 2021, y permitan valorar si son suficientemente buenos como para iniciar un ensayo de fase 3. En esa fase se encuentra ya otra terapia celular (NurOwn) a base de células madre mesenquimales que había tenido resultados aparentemente positivos en los ensayos de fase 1/2 y fase 2a, y de la que se han publicado resultados preliminares de seguridad en fase 3 en 31 pacientes; pero es importante tener todos los datos completos del ensayo en fase 3 antes de poder estar seguros de su eficacia.
En definitiva, según lo expuesto hasta aquí, aunque se están pasos en la buena dirección, muchos de los avances en potenciales terapias requieren aún de su estudio en pacientes con ELA y otros están en sus fases iniciales de evaluación en humanos, por lo que los resultados deben ser interpretados con cautela hasta que se tengan datos robustos de ensayos clínicos.
La ELA y, en general, todas las patologías neurológicas degenerativas –aún sin cura– tienen una extraordinaria importancia clínica y un enorme impacto socioeconómico, que desborda los datos meramente estadísticos, dadas sus implicaciones personales y sociales. Por ello, el papel del farmacéutico como profesional sanitario tiene el múltiple cometido de colaborar activamente tanto en la detección precoz como en el proceso terapéutico, así como en la atención necesaria que los pacientes y sus cuidadores requieren. Este último aspecto social es especialmente relevante, habida cuenta del elevado grado de dependencia de los pacientes, que no se limita a las fases más avanzadas de la enfermedad. Al desgaste físico que impone la atención más inmediata, junto con el tiempo dedicado a ésta, en los cuidadores se une otro desgaste de peores consecuencias, el emocional, que en el caso de la ELA puede llegar a ser especialmente grave.
Dadas las particularidades de la enfermedad, todos los pacientes con diagnóstico de ELA van a estar en tratamiento crónico mayoritariamente en el ámbito ambulatorio (pudiendo requerir ingresos hospitalarios frecuentes) y, con seguridad, van a ser tratados, además de con riluzol, con más de un fármaco para manejar los síntomas más molestos, siendo la adherencia terapéutica un pilar fundamental con influencia en la calidad de vida de los pacientes. En ese contexto, la proximidad y accesibilidad del farmacéutico para los pacientes permite que pueda ejercer una labor asistencial proactiva a través de los Servicios Profesionales Farmacéuticos Asistenciales.
Participando del equipo multidisciplinar necesario en el abordaje individualizado de un paciente con ELA, el farmacéutico comunitario, en colaboración con los especialistas del ámbito hospitalario, participará decisivamente en la consecución de los objetivos terapéuticos, facilitando la disponibilidad de los medicamentos usados en el tratamiento sintomático, con claras implicaciones en la sostenibilidad del Sistema Nacional de Salud. Además, el hecho de que la oficina de farmacia sea el establecimiento sanitario más accesible (con amplitud de horarios y sin necesidad de cita previa), ubicuo y cercano para los pacientes, posiciona a la red española de 22.000 farmacias como un privilegiado centro de divulgación de información rigurosa y de promoción de un mejor uso de los medicamentos, previniendo los problemas relacionados con los mismos.
Se describen, a continuación, las principales vías asistenciales de actuación del profesional farmacéutico para con los pacientes con ELA y sus cuidadores.
Dado que las necesidades de los pacientes se multiplican con la evolución de la enfermedad, la atención temprana contribuye a su mejor formación y a la de sus cuidadores para prevenir situaciones de deterioro y afrontar el proceso de dependencia. El primer punto que debe tener claro la familia, y así debe transmitírselo el farmacéutico, es que la ELA es una enfermedad neurodegenerativa para la que aún no se ha encontrado curación ni tratamiento que la frene de forma eficaz.
Hay que subrayar que el tratamiento, sintomático y paliativo, va dirigido a retrasar la evolución de la enfermedad, conservando –en lo posible– las capacidades cotidianas del paciente y mejorando su calidad de vida. Se debe, por tanto, desacreditar las terapias “milagrosas”, que individuos o empresas puedan anunciar, generalmente a través de internet, con el fin de manipular emocional y económicamente a los pacientes y sus familias, ofreciendo garantías de unos resultados que en ningún caso van a conseguir, además de ser ilegales en la Unión Europea.
Adicionalmente, el farmacéutico deberá facilitar a los pacientes y cuidadores recomendaciones para paliar los síntomas que van apareciendo (Figura 5):
A medida que la ELA progresa, será necesaria la utilización de algunas ayudas técnicas (incluyendo dispositivos, instrumentos, equipos informáticos) para proteger, paliar o sustituir funciones, prevenir deficiencias o limitaciones funcionales crecientes. Será necesaria la adaptación del entorno del paciente, de manera que se facilite su movilidad siempre manteniendo ciertas medidas de seguridad para evitar accidentes y complicaciones; por ejemplo, con el fin de evitar caídas, conviene retirar o fijar alfombras y cables, evitar la colocación de muebles que obstaculicen las vías de paso, etc. Desde la farmacia comunitaria se pueden facilitar al cuidador catálogos y empresas distribuidoras de dispositivos y ayudas técnicas (algunas pueden dispensarse en la propia farmacia, especialmente si dispone de Ortopedia).
Muchos de los dispositivos no están diseñados específicamente para pacientes con ELA (se usan los ya comercializados para otras patologías y se adaptan a las necesidades de los pacientes), por lo que no siempre su utilización será un éxito. Las ayudas técnicas más usadas son: cubiertos adaptados y vasos con tetina para ayudar a la alimentación, inspirómetros para realizar ejercicios de respiración, humidificadores, camas articuladas y grúas de transferencia, colchón anti-escaras, sillas de ruedas o de ducha, elevadores para el inodoro, asideras para la sujeción en el baño, muletas, bastones o andadores, collarines cervicales o avisadores acústicos. También se podrán recomendar productos sanitarios como: apósitos protectores para disminuir las úlceras cutáneas por el uso de mascarillas, gasas y esparadrapos antialérgicos para tapar la sonda nasogástrica, pañales absorbentes, colectores de orina o sondas de incontinencia, esponjas jabonosas para el aseo en la cama, etc.
Llegado el caso, la recomendación de tecnologías y ayudas para la comunicación puede contemplar el uso de dispositivos –eléctricos o con baterías– muy diversos: a) amplificadores de voz (micrófonos): dispositivos portátiles que pueden asentarse en la cabeza, la mano o apoyarse en la ropa, y aumentan el volumen de la voz, si la dicción es clara pero de tono insuficiente; b) ayudas a la comunicación de expresión de la voz (VOCAs): expresan oralmente un texto escrito y señalado con el dedo o, en los más avanzados, con un teclado, un ratón adaptado, un mando tipo joystick o hasta con un trazado ocular (lo más moderno y útil, pues la mayoría de pacientes conserva la movilidad ocular); c) sistemas basados en ordenadores: programas muy variados, tanto en uso como en estructura, disponibles para todo tipo de dispositivos (ordenadores, tabletas, teléfonos, etc.), que permiten múltiples funcionalidades e individualización (emisión de voz, de palabras concretas); y d) programas de escritura: suelen incorporase en ordenadores y teléfonos y, ante la dificultad del manejo de los dedos, pueden ayudar a transformar la dicción en escritura para comunicaciones tipo correos electrónicos o textos elaborados por el usuario.
Por último, debe recordársele al cuidador que, especialmente durante las fases avanzadas de la enfermedad, algunos pacientes pueden padecer ciertas formas de demencia, lo que aconseja mantener el contacto visual, evitando colocarse demasiado lejos o demasiado cerca del paciente, recurriendo al contacto físico cuando sea preciso para mantener la atención. En cualquier caso, debido a las notables limitaciones de comunicación que presentan, es muy importante no tener prisa en las respuestas y nunca mostrarse agresivo.
La importancia de un diagnóstico temprano se debe a que puede incrementar la probabilidad de iniciar precozmente las terapias neuroprotectoras que puedan minimizar el deterioro neuronal y prolongar la supervivencia, especialmente en los pacientes menores de 50 años. Asimismo, facilita el establecimiento de tratamientos sintomáticos que mejoran la calidad de vida del paciente y permite prolongar su autonomía en las actividades cotidianas. Pero lo cierto es que la detección precoz de la enfermedad sigue siendo muy dificultosa y el tiempo entre la aparición de los primeros síntomas hasta el diagnóstico se prolonga durante meses; como ya se ha indicado, se debe al menos en parte a la falta de familiaridad de los profesionales sanitarios con la enfermedad, la presentación con formas inusuales, la coexistencia de otra enfermedad o los falsos negativos ante la existencia de hallazgos neurorradiológicos que no justifican la ELA.
Por ello, es importante que el farmacéutico comunitario sepa detectar síntomas y signos de alarma que pudieran presentar algunas personas y, en su caso, derivar al médico a fin de confirmar o descartar cualquier sospecha inicial. El estudio específico y diagnóstico del paciente candidato a padecer ELA está reservado al médico especialista, específicamente el neurólogo. Dado el carácter multifactorial de la enfermedad y la diversidad de cuidados que requieren los pacientes, la colaboración coordinada de los diferentes profesionales sanitarios resulta aún más imprescindible de lo habitual, ya que tanto en el diagnóstico como en las evaluaciones periódicas participarán un gran número de profesionales.
Los signos y síntomas de alarma que podemos observar y por los que debe derivarse al médico a cualquier persona que acuda a la farmacia comunitaria y diga experimentarlos desde hace un tiempo se recogen en la Figura 6. Raramente los pacientes referirán signos de atrofia o fasciculaciones, pues no saben verlas. Para ello es necesario observar un grupo de músculos de las extremidades, tórax o abdomen detenidamente y se podrán observar las contracciones musculares.
Una vez establecido el diagnóstico y el tratamiento por el neurólogo, debe mantenerse una estricta coordinación con éste para evitar dar mensajes discordantes al cuidador. Dentro de los servicios profesionales farmacéuticos asistenciales, en el caso del tratamiento de la ELA cobran especial protagonismo la dispensación, la indicación farmacéutica y el seguimiento farmacoterapéutico, sin olvidarnos de la adherencia terapéutica y la farmacovigilancia.
Según se ha indicado, el único fármaco neuromodulador de la enfermedad, el riluzol, es de dispensación hospitalaria, pero todos aquellos fármacos para tratar los diferentes síntomas de la enfermedad son dispensados desde las farmacias comunitarias. Durante la dispensación, el farmacéutico debe evaluar que la prescripción del medicamento se corresponde con las necesidades del paciente, verificando que no existen condiciones para la no dispensación del mismo. Según va avanzando la enfermedad, debido a las condiciones del paciente, es frecuente que un familiar próximo o cuidador se encargue de recoger la medicación.
Además de los medicamentos sujetos a prescripción médica, en muchas ocasiones el propio paciente o el cuidador solicitará consejo al farmacéutico sobre los diferentes síntomas que van apareciendo. En ese caso, mediante el servicio de indicación, el farmacéutico podrá recomendar espesantes para aumentar la consistencia de los alimentos y evitar atragantamientos, o suplementos nutricionales orales para conseguir alcanzar los requerimientos calóricos necesarios cuando la ingesta oral es insuficiente. Otros productos de indicación farmacéutica pueden ser la fibra, complementos alimenticios o vitamina D para paliar los primeros síntomas musculares. Aunque, en general, se acepta que salvo expresa prescripción por el médico responsable del paciente, normalmente no se requiere ningún tipo de suplemento vitamínico o nutricional.
— El farmacéutico contribuye eficazmente en la asistencia sanitaria individualizada a los pacientes con ELA—
Es importante subrayar que el paciente con ELA suele estar polimedicado y presentar otras comorbilidades, por lo que será muy importante analizar el perfil de todos los medicamentos prescritos por el médico. Así, el servicio de seguimiento farmacoterapéutico (SFT) puede ser muy útil para detectar y prevenir problemas relacionados con los medicamentos (PRM) y resolver resultados negativos asociados a la medicación (RNM). Durante este servicio se deberán evaluar los diferentes signos y síntomas (presencia de sialorrea, disfagia, pérdida de peso, alteraciones del lenguaje, estreñimiento, etc.) de la enfermedad para ir viendo la evolución y colaborar con el médico en la instauración de tratamiento para nuevos síntomas que vayan apareciendo o cambios de las pautas terapéuticas establecidas; además, se debe prestar atención a otras demandas que los pacientes y cuidadores puedan tener (por la confianza con el farmacéutico). Para una mayor información sobre el SFT en pacientes con ELA, se recomienda consultar la publicación de López-Casero y colaboradores (2019).
Por la situación de polimedicación de los pacientes, desde la farmacia comunitaria también resulta especialmente interesante ofrecerles (o al cuidador) un servicio personalizado de dispensación (SPD), empleando para ello dispositivos adecuados para una correcta aplicación de las pautas posológicas –en ocasiones complicadas– y una adecuada adherencia al tratamiento de los medicamentos prescritos por el médico. Resulta muy útil proporcionar al cuidador las instrucciones de la medicación y su pauta por escrito, siempre de la forma más sencilla y, en la medida de lo posible, bajo la forma de lista o rutina diaria. Dado el caso, es importante orientar la intervención realizada por el farmacéutico hacia la causa específica que origina falta de adherencia, de manera personalizada para que se ajuste a cada paciente; en estadios avanzados de la ELA, la falta de adherencia se relaciona con la disfagia, y puede plantearse adala adaptación de las formas farmacéuticas sólidas y convertirlas en preparaciones de fácil deglución (en aquellas en las que se pueda).
Durante el proceso terapéutico, el farmacéutico deberá asegurarse de que el paciente se involucra voluntariamente en su tratamiento y toma la medicación de manera correcta (ya que únicamente así será efectivo), actuando como agente centinela ante posibles reacciones adversas12 , contraindicaciones o interacciones farmacológicas que puedan emerger con cualquier medicamento, siendo fundamental evitar el consumo de medicamentos que no hayan sido prescritos por el médico (aunque sean de uso habitual).
A modo de recordatorio, el único fármaco autorizado para el tratamiento de la ELA, el riluzol, no es especialmente tóxico, siendo raros los efectos adversos graves. No obstante, en la evaluación de los pacientes debe tenerse en cuenta que algunos efectos adversos menos relevantes clínicamente pueden ser bastante comunes: la fatiga, la astenia, las náuseas y una elevación de los niveles de transaminasas; con menor frecuencia (1‐10%) pueden aparecer diarrea, dolor abdominal, vómitos, cefalea, mareos, parestesia, somnolencia y dolor.
De forma concomitante, es especialmente frecuente el uso de amitriptilina, que presenta unos marcados efectos anticolinérgicos (sequedad permanente de boca, taquicardia, estreñimiento, etc.), o de antidepresivos ISRS, como el citalopram o la fluoxetina (que suelen caracterizarse por molestias digestivas, cefalea, etc.). Solo es aconsejable el uso de agentes mucolíticos, como la acetilcisteína, cuando la capacidad de toser del paciente no esté muy mermada. Es común, especialmente durante las fases más avanzadas, el uso de agentes hipnóticos, que pueden provocar somnolencia durante el día (por ello, se suele preferir el empleo de agentes de duración corta, como el zolpidem). Los antisecretores gástricos, como la ranitidina o el omeprazol, son de uso habitual para reducir las potenciales consecuencias de un reflujo gastroesofágico. También se hace habitual el empleo de laxantes, para tratar el extreñimiento. Para una mayor información sobre reacciones adversas, contraindicaciones y potenciales interacciones de todos estos fármacos, se recomienda consultar fuentes especializadas, como la base de datos del conocimiento sanitario BOT PLUS.
Al ser la ELA una enfermedad que genera dependencia, produce en los afectados y/o sus cuidadores una sensación de soledad y de exclusión social. El soporte económico, sanitario, social y emocional por parte de los servicios sociales públicos y las asociaciones de pacientes son fundamentales en el proceso de la enfermedad. Desde la farmacia es importante aconsejar al cuidador sobre la conveniencia de integrarse en asociaciones de pacientes, que pueden ayudar en gran manera a la hora de conseguir un asesoramiento personalizado sobre las ayudas oficiales disponibles para hacer frente a los siempre onerosos cuidados requeridos por los pacientes. Las funciones sociales de información, orientación al ciudadano, colaboración en iniciativas sociales de promoción y organización de voluntariado, etc. deben orientarse a reducir esa sensación de exclusión social.
Tal y como se ha venido exponiendo, la figura del cuidador (reconocida Ley 39/2006, de Promoción de la Autonomía Personal y Atención a las personas en situación de Dependencia) es imprescindible para los enfermos de ELA: es el informador clave, quien supervisa, provee los cuidados y administra los tratamientos prescritos, y quien participa activamente en la toma de decisiones. Pero, hoy en día, en España, solo pocas familias (un 6% de los casos) se pueden permitir contratar a cuidadores externos y es un familiar quien la asume. A pesar de que la mayor parte de los afectados de ELA presentan un elevado grado de discapacidad física, solo un tercio dispone de un único cuidador, que normalmente es una mujer (75%), habitualmente la pareja, hijas o madres. O sea, en la mayoría de casos en nuestro país, es un familiar no profesional quien se encarga de los cuidados de los afectados de ELA.
Resulta necesario, por tanto, vigilar la sobrecarga del cuidador principal, para lo cual el farmacéutico podrá recomendarle ciertas pautas:
Finalmente, el papel del farmacéutico resulta clave en la orientación a pacientes y cuidadores y los consejos sobre dónde acudir cuando son diagnosticados. En ese sentido, aunque la ELA debe atenderse en los servicios de neurología comunitarios, es importante la existencia de unidades especializadas de ELA para, entre otras funciones, garantizar una segunda opinión, que evite el retraso diagnóstico en casos complejos y pueda ofrecer o asesorar en la oferta de técnicas de tratamiento. Esas unidades especializadas suelen estar compuestas por un equipo experto de neurólogos que coordina al resto de especialidades que participan en el seguimiento/tratamiento del paciente: neumología, cardiología, rehabilitación, traumatología, otorrinolaringología, nutrición, genética, fisioterapia, servicios sociales, psiquiatría etc. Habitualmente compaginan la labor asistencial con la docencia y la investigación. Todos los especialistas evalúan al paciente en régimen de cita única y en el mismo espacio, registrando su evaluación en una hoja común de la historia clínica para evitar evaluaciones reiterativas al paciente y así emitir un informe único.
En 2006 se creó de manera oficial la Unidad de ELA y Otras Enfermedades de la Motoneurona del Hospital Carlos III, la primera Unidad en España. Desde entonces, se han creado varias más, cuyo listado puede consultarse en la página web de la Asociación Española de la ELA (https://adelaweb.org/la-ela/unidades-de-ela/). El centro que clásicamente ha centralizado los estudios de pacientes con ELA Familiar en España se encuentra en la Unidad de ELA del Hospital 12 de Octubre, que desde el año 2006 ha pasado a constituir el Laboratorio de Investigación en ELA del Instituto de Investigación Sanitaria del Hospital 12 de Octubre “i+12”.
Las ya mencionadas organizaciones de lucha contra la ELA juegan un papel muy importante en la orientación y apoyo a las personas enfermas y sus familiares, así como en el fomento de cualquier investigación relacionada con la patología (terapéutica, genética, farmacológica, etc.). Ayudarán a los pacientes a orientarse dentro de la complicada situación que están viviendo e informarse sobre los recursos, ayudas y servicios disponibles cerca de su lugar de residencia. Ofertan, además, una amplia cartera de servicios, a domicilio y en sus propias sedes, como psicología, fisioterapia, logopedia, trabajo social, banco de ayudas o préstamo de productos de apoyo para la autonomía o voluntariado. También informan sobre los avances en investigación y ensayos clínicos en marcha. En la actualidad, existen varias entidades de ámbito nacional y otras a nivel autonómico que prestan estos servicios a personas con ELA y a sus familiares, y se recogen en la Tabla 2.
El cáncer de páncreas es uno de los tumores humanos con un peor pronóstico: la tasa de supervivencia a los 5 años se sitúa por debajo del 10%. La ausencia de mejoras terapéuticas en los últimos tiempos ha determinado que la supervivencia de los pacientes no mejore. Además, el aumento de su incidencia lo ha situado como una causa relevante de mortalidad: en España, hubo más muertes por cáncer de páncreas que por cáncer de mama en 2018, siendo la tercera causa de muerte por cáncer. La quimioterapia convencional ha mostrado escasa eficacia, que no ha sido mejorada por la más moderna inmunoterapia; de hecho, los inhibidores de la proteína de muerte celular programada 1 (PD-1), como pembrolizumab o nivolumab, que en los últimos años han sido autorizados frente a múltiples tipos de tumores, solo tienen un efecto limitado en el adenocarcinoma ductal pancreático (ADP), lo que subraya la necesidad buscar vías alternativas de tratamiento.
Un grupo de investigadores, con participación española, planteó la posibilidad de combinar esa acción farmacológica con el bloqueo del receptor 4 de la quimiocina CXC (CXCR4), que promueve la infiltración de células T en los tumores y es sinérgico con la terapia anti-PD-1 en modelos de ratón con ADP. Así, un reciente ensayo clínico multicéntrico de fase 2a (COMBAT), no aleatorizado y abierto, de dos cohortes, evaluó la seguridad, la eficacia y la farmacodinamia de la combinación del antagonista de CXCR4 motixafortida (BL-8040, designado en enero del 2020 como medicamento huérfano por la EMA) y pembrolizumab, con o sin quimioterapia, en ADP metastásico e inoperable. La variable primaria de eficacia fue la tasa de respuesta objetiva (TRO), determinándose como variables secundarias la supervivencia general (SG), la tasa de control de la enfermedad (TCE) y la seguridad.
En la cohorte 1 se incluyeron 37 pacientes con enfermedad resistente a quimioterapia, quienes recibieron motixafortida y pembrolizumab. La tasa de control de la enfermedad fue del 34,5% en la población evaluable (N= 29): 9 pacientes (31%) con enfermedad estable y 1 paciente (3,4%) con respuesta parcial; la mediana de la SG fue de 3,3 meses en la población ITT (por intención de tratar). De forma interesante, en el subgrupo de pacientes que recibieron la combinación en estudio como terapia de segunda línea, la mediana de la SG crecía hasta los 7,5 meses. En la cohorte 2, 22 pacientes recibieron motixafortida y pembrolizumab junto con quimioterapia (irinotecán, fluorouracilo y ácido folínico); se obtuvo una TRO del 32%, una tasa de control de la enfermedad del 77% y una mediana de duración de respuesta (SG) de 7,8 meses, respectivamente. Esto supone un aumento de la supervivencia de 4,5 meses frente al uso de inmunoterapia sola. Además, se demostró que el nuevo fármaco, motixafortida, aumentaba la infiltración tumoral de células T efectoras CD8+ y disminuía las células supresoras mieloides y las células T reguladoras circulantes (estas últimas en mayor medida).
Tales resultados –sobre todo, los de la cohorte 2– sugieren, a pesar de las limitaciones del estudio (fundamentalmente, su tamaño), que el bloqueo combinado de CXCR4 y PD-1 puede ampliar el beneficio de la quimioterapia en adenocarcinoma ductal pancreático. Si bien se debe garantizar la confirmación en ensayos aleatorizados y controlados más amplios, se trata de la primera noticia positiva en mucho tiempo, que plantea una estrategia potencialmente beneficiosa –combinación de inmunoterapia y quimioterapia– en cáncer de páncreas. Los resultados invitan a creer que la barrera de la inmunoterapia en este tipo de tumor puede ser superada en un futuro a medio plazo.
Bien es sabido a estas alturas de año que el nuevo coronavirus SARS-CoV-2 se transmite de persona a persona fundamentalmente (y, quizás, exclusivamente) a través del contacto directo mediante gotículas respiratorias emitidas al hablar, toser o estornudar, por una persona infectada. Con el objetivo de analizar la evidencia disponible sobre los efectos de la distancia física, el uso de mascarillas y de protección ocular en la prevención de la transmisión del virus en entornos de centros sanitarios y en la comunidad, un equipo internacional de investigadores ha llevado a cabo revisión sistemática y un meta-análisis en que incluyeron también estudios que analizaban otros betacoronavirus, como el SARS-CoV y el MERS-CoV.
Para ello, realizaron una búsqueda en 21 fuentes bibliográficas específicas de la OMS y de la COVID-19 hasta el 3 de mayo de 2020, sin restricción por idioma. Examinaron los registros y extrajeron los datos para realizar un meta-análisis bayesiano y una meta-regresión de efectos aleatorios; la calidad de la evidencia se calificó de acuerdo con los métodos de Cochrane y el enfoque GRADE. Tras dicha búsqueda, identificaron 172 estudios observacionales en 16 países y 5 continentes, con 44 estudios comparativos relevantes en entornos de atención médica y no médica (N= 25.697 pacientes), pero sin ningún ensayos controlados y aleatorizados.
Los resultados de este trabajo demuestran que la transmisión de coronavirus es menor con un distanciamiento físico de 1 m o más en comparación con una distancia de < 1 m (N= 10.736), siendo el cociente de probabilidad ajustado (ORa) de 0,18 (IC95% 0,09 a 0,38) y la diferencia de riesgo (DR) de -10,2% (IC95% -11,5 a -7,5) (evidencia de calidad moderada): esto se traduce en que, con una separación física de menos de 1 m hasta el paciente infectado, se contagiarían hasta el 12,8% de las personas expuestas, porcentaje que se reduce al 2,6% si se mantiene una distancia superior a 1 m. Además, la protección frente al contagio se incrementa a medida que se alarga la distancia, con un cambio el riesgo relativo (RR) de 2,02 puntos porcentuales por metro (p= 0,041; evidencia de calidad moderada). Por otro lado, una evidencia de calidad baja respalda que el uso de mascarillas (N= 2.647) puede también resultar en una importante reducción del riesgo de infección con un ORa de 0,15 (IC95% 0,07 a 0,34) y una DR de -14,3% (17,4% de contagios vs. 3,1% sin mascarillas) (IC95% -15,7 a -10,7); el efecto en la reducción del riesgo es más notable con las mascarillas N95 o similares (FFP2 o KN95) en comparación con el uso de mascarillas quirúrgicas desechables o higiénicas (por ejemplo, mascarillas de algodón reutilizables de 12-16 capas) (p= 0,090; evidencia de calidad baja). Finalmente, la protección ocular (N= 3.713) también se asoció con un menor riesgo de infección, con un ORa de 0,22 (IC95% 0,12 a 0,39) y una DR de -10,6% (IC95% -12,5 a -7,7) (baja calidad de la evidencia). Los estudios no ajustados y los análisis de subgrupos y sensibilidad mostraron hallazgos similares.
En resumen, este artículo representa la primera gran revisión de la evidencia disponible y refrenda que el distanciamiento físico entre personas de al menos 1 metro, el uso de mascarillas filtrantes y la protección ocular (esta medida, muchas veces subestimada) son medidas profilácticas eficaces para evitar la propagación de la COVID-19 tanto en el entorno de atención sanitaria como en la comunidad. Proporciona estimaciones cuantitativas que pueden apoyar y justificar posibles políticas de salud pública. No obstante, los propios autores indican que se necesitan ensayos aleatorizados sólidos que aporten una evidencia de mayor calidad sobre dichas intervenciones, si bien los resultados comentados pueden servir para la toma de medidas provisionales.

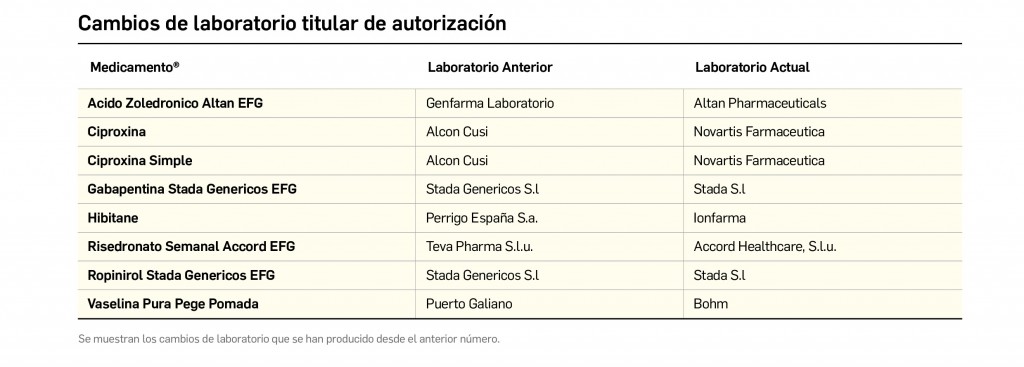
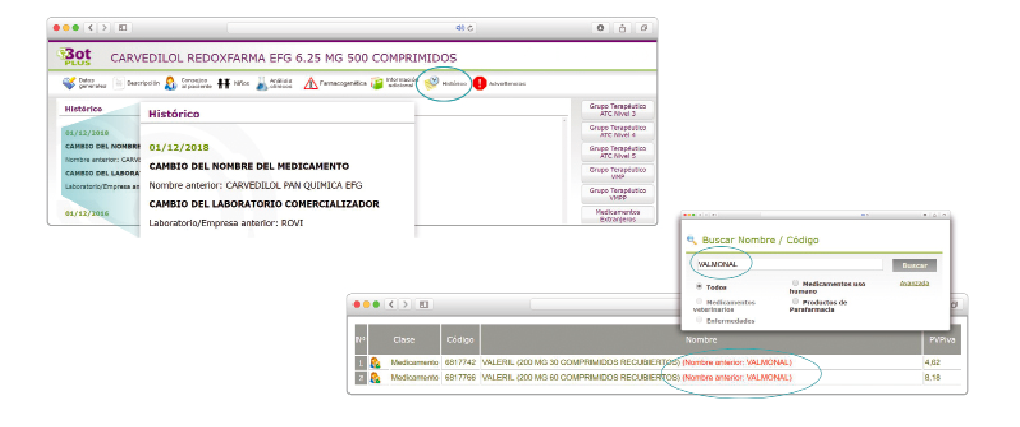
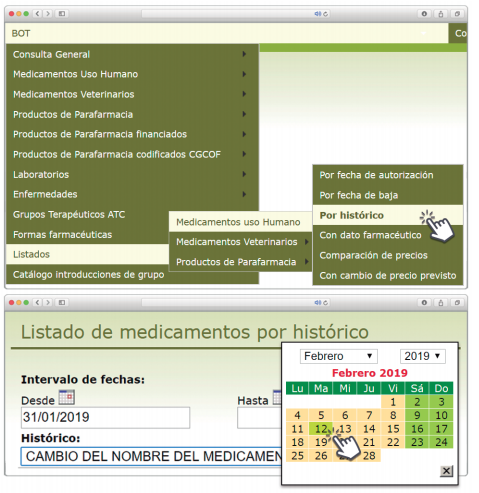
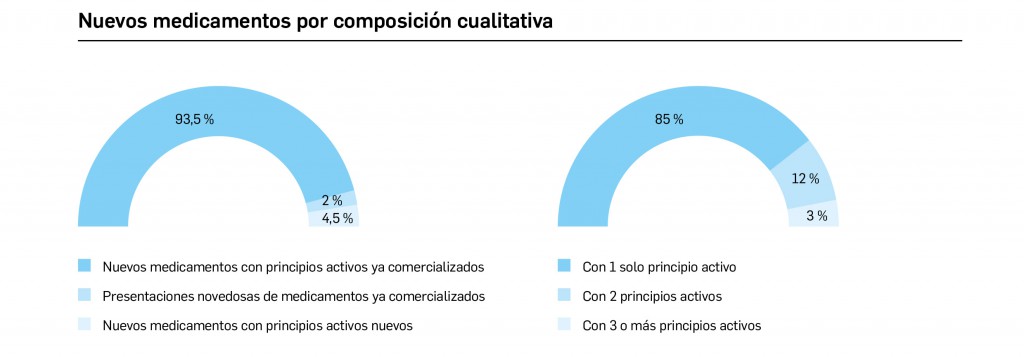
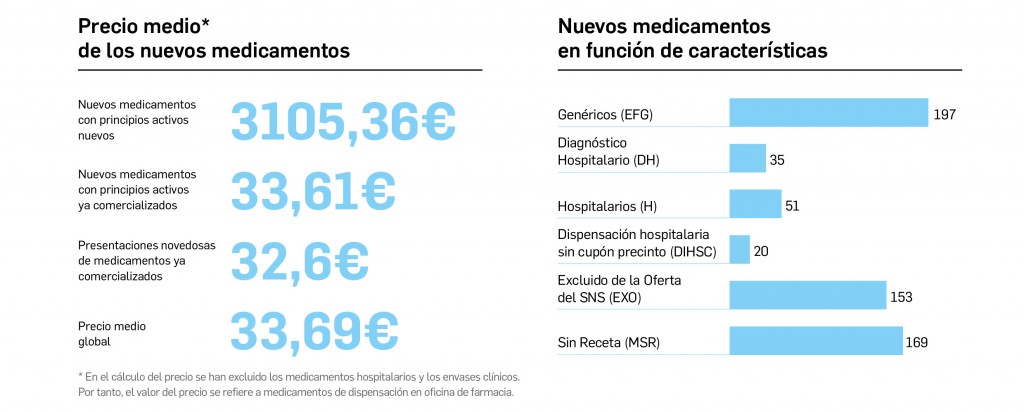
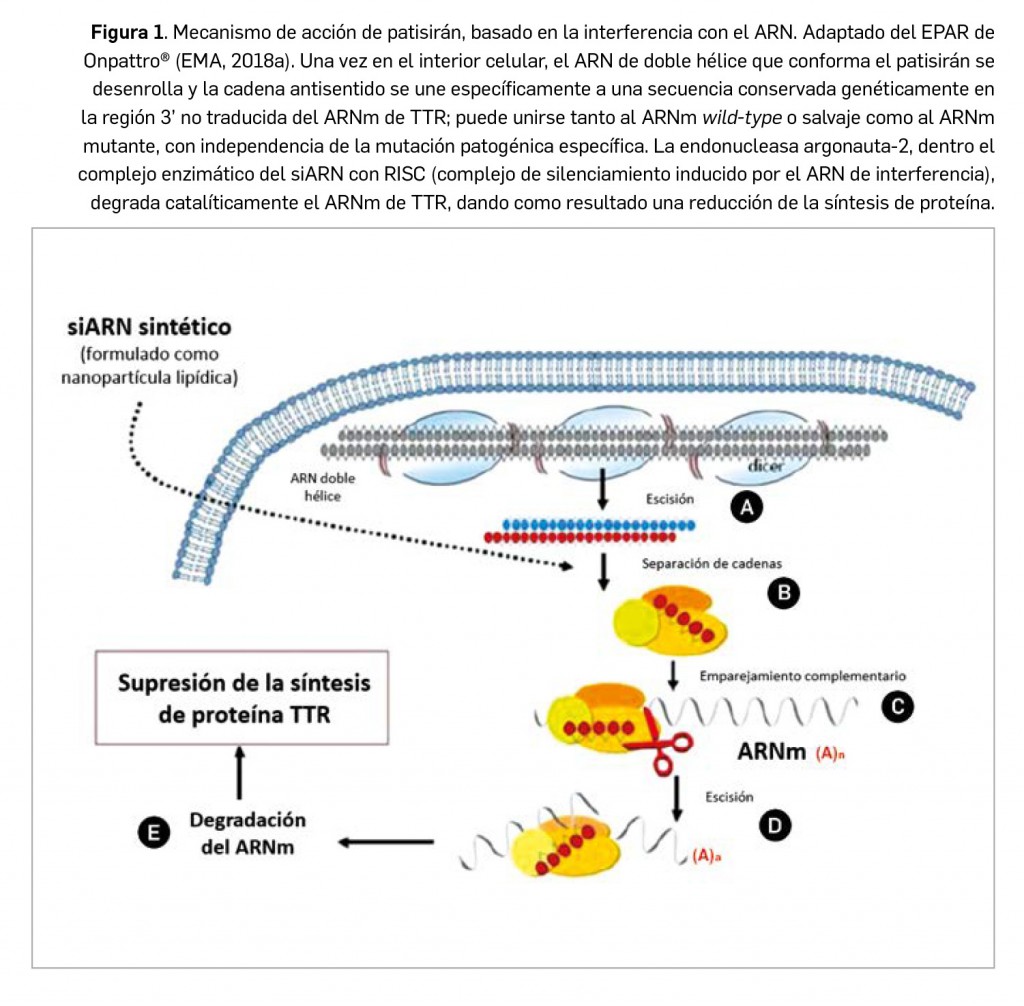
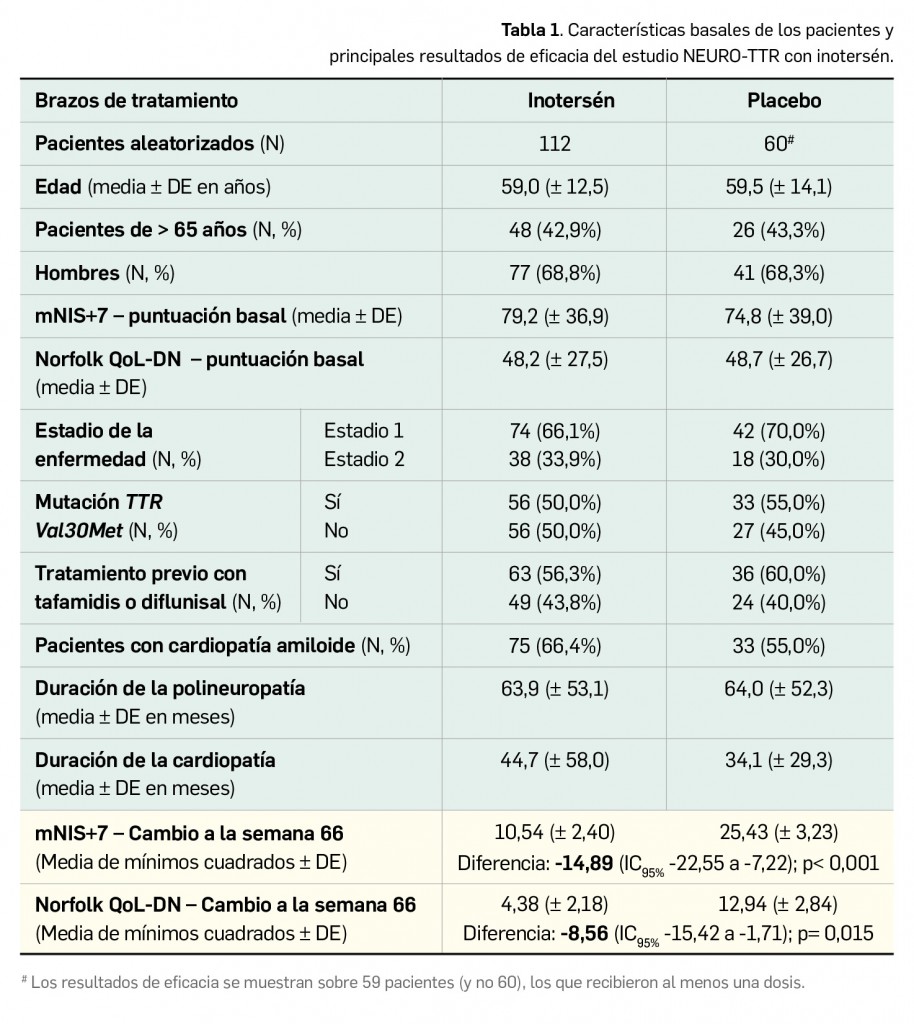
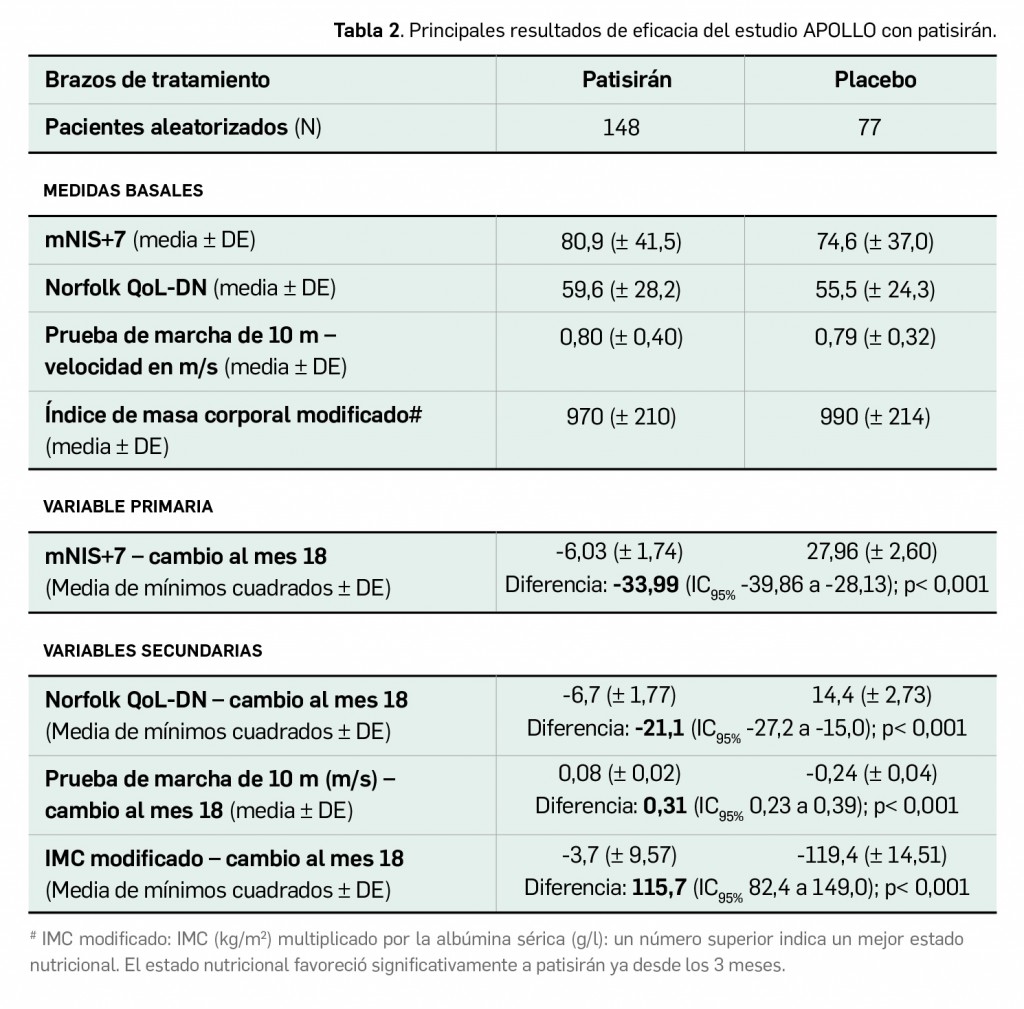



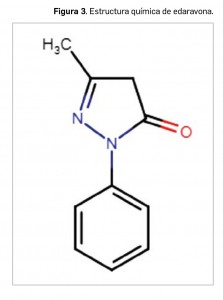
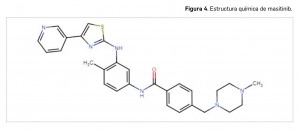
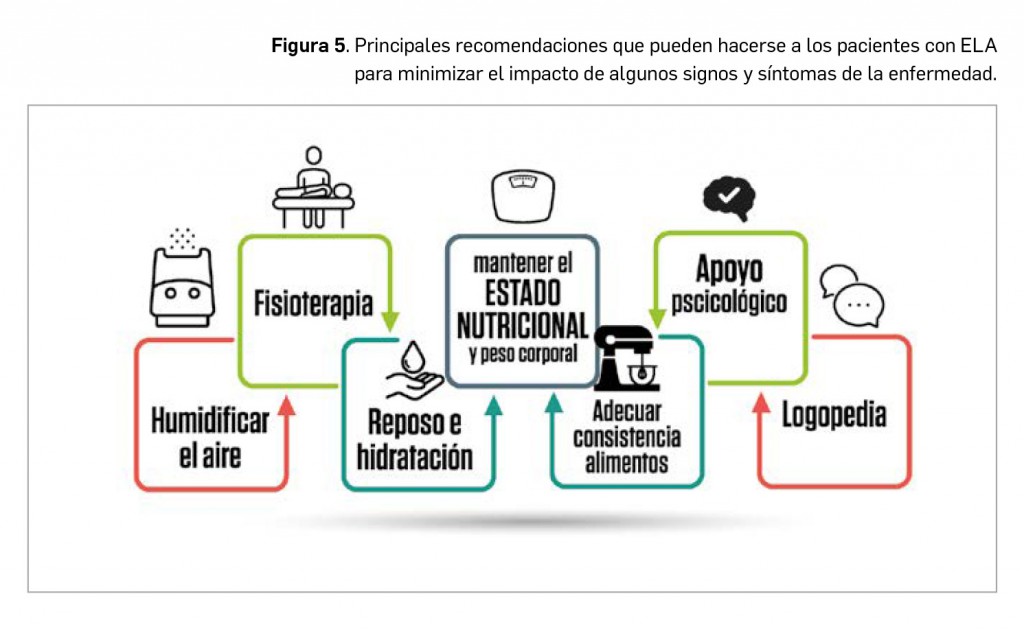
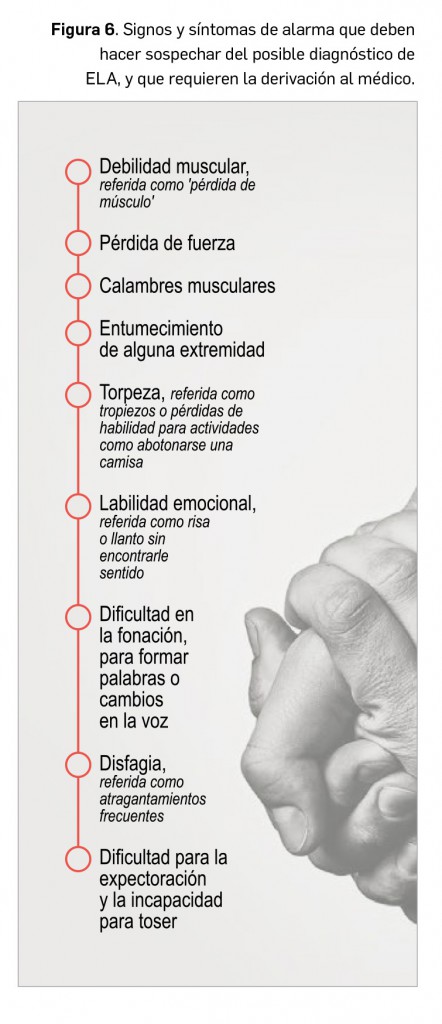
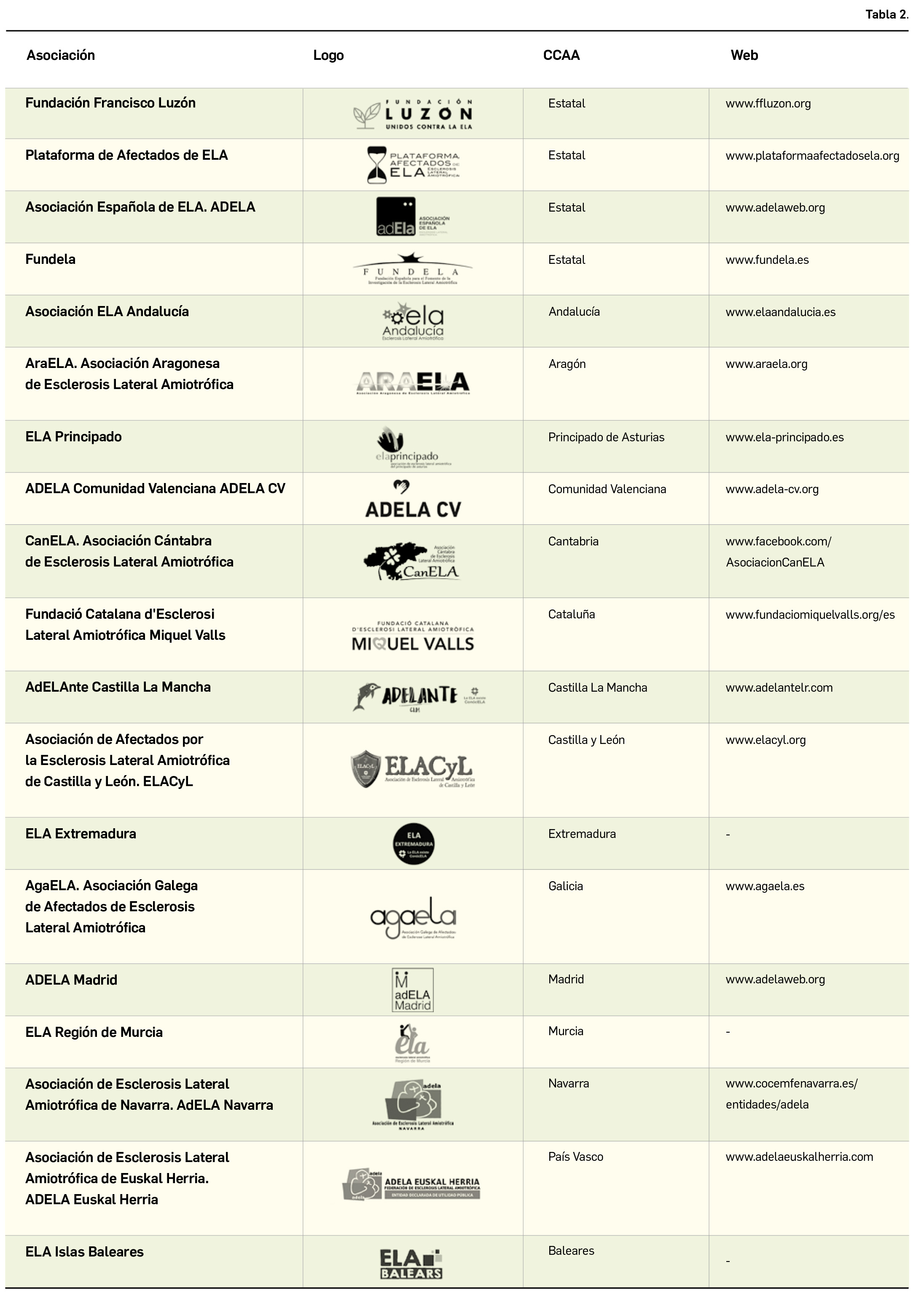
{kind=link}
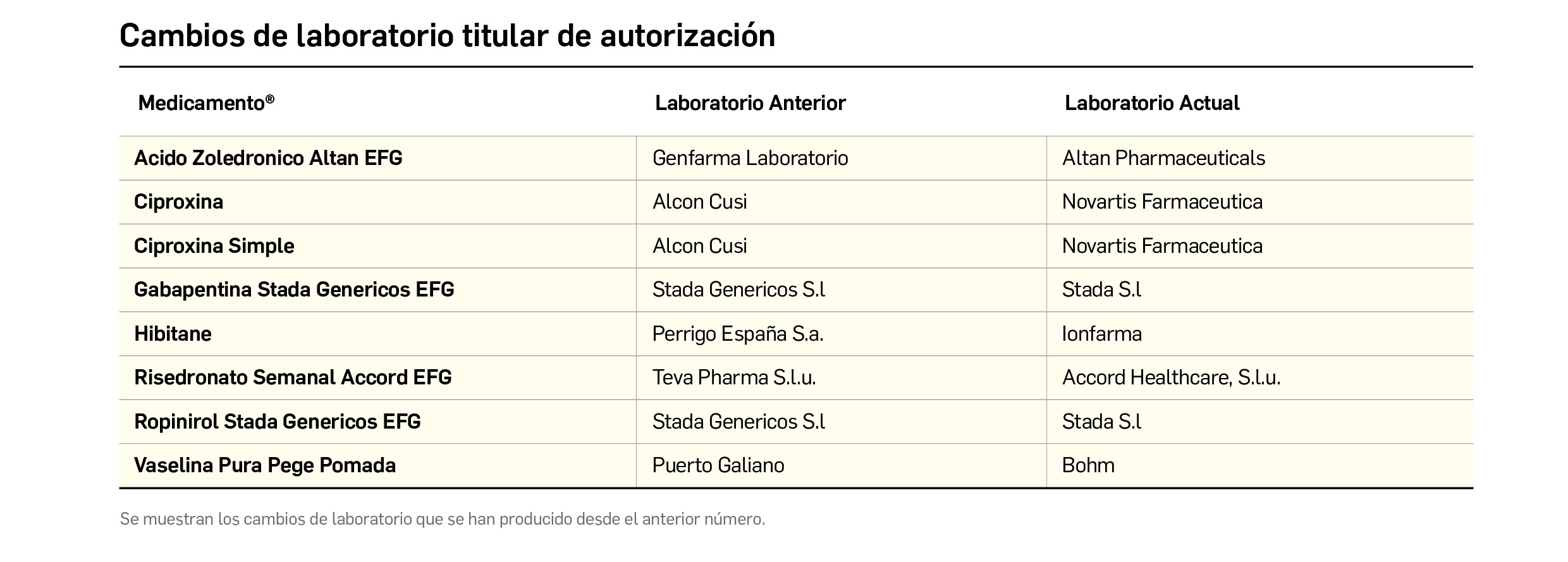{kind=link}
{kind=link}
{kind=link}
{kind=link}
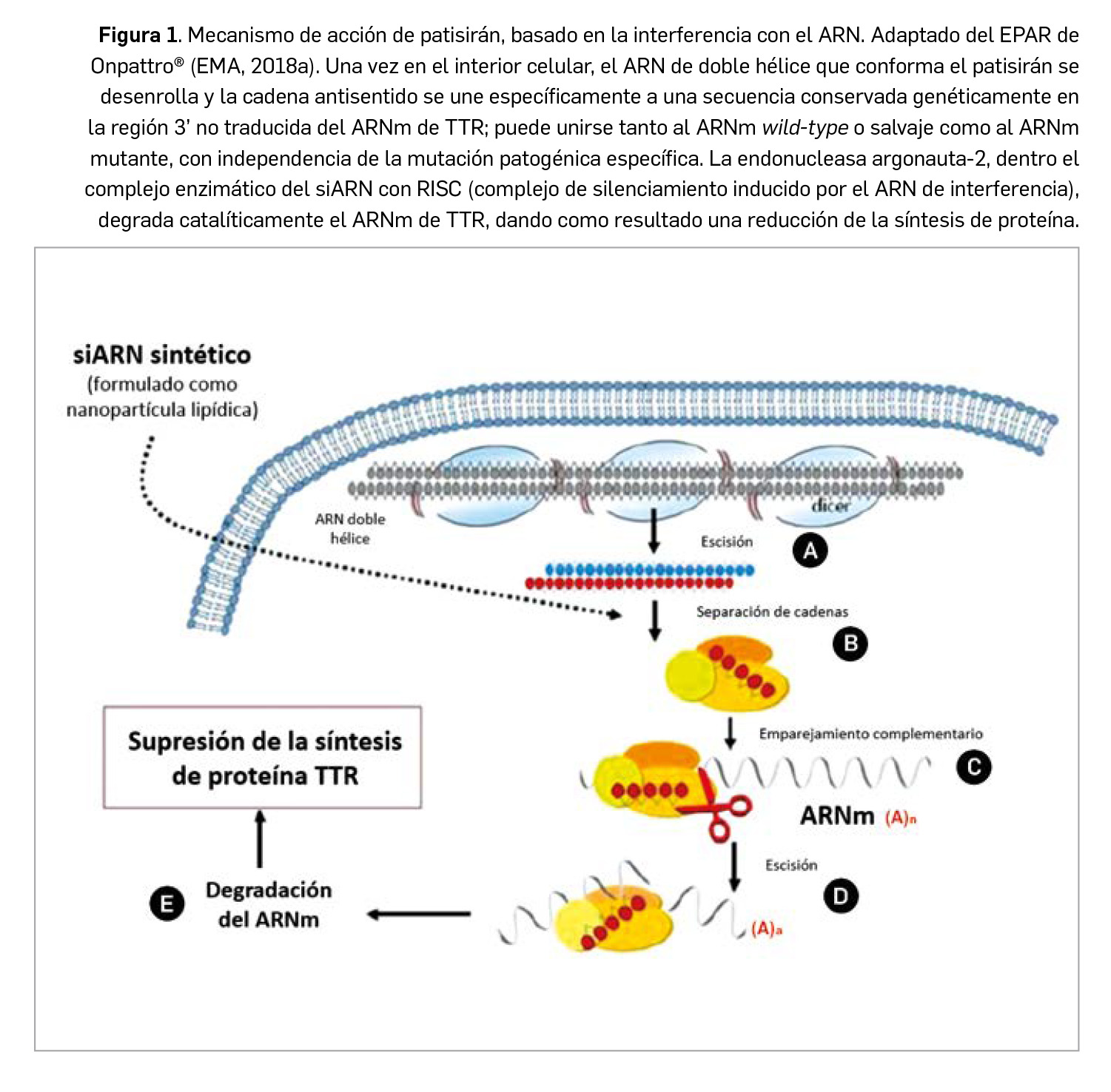{kind=link}
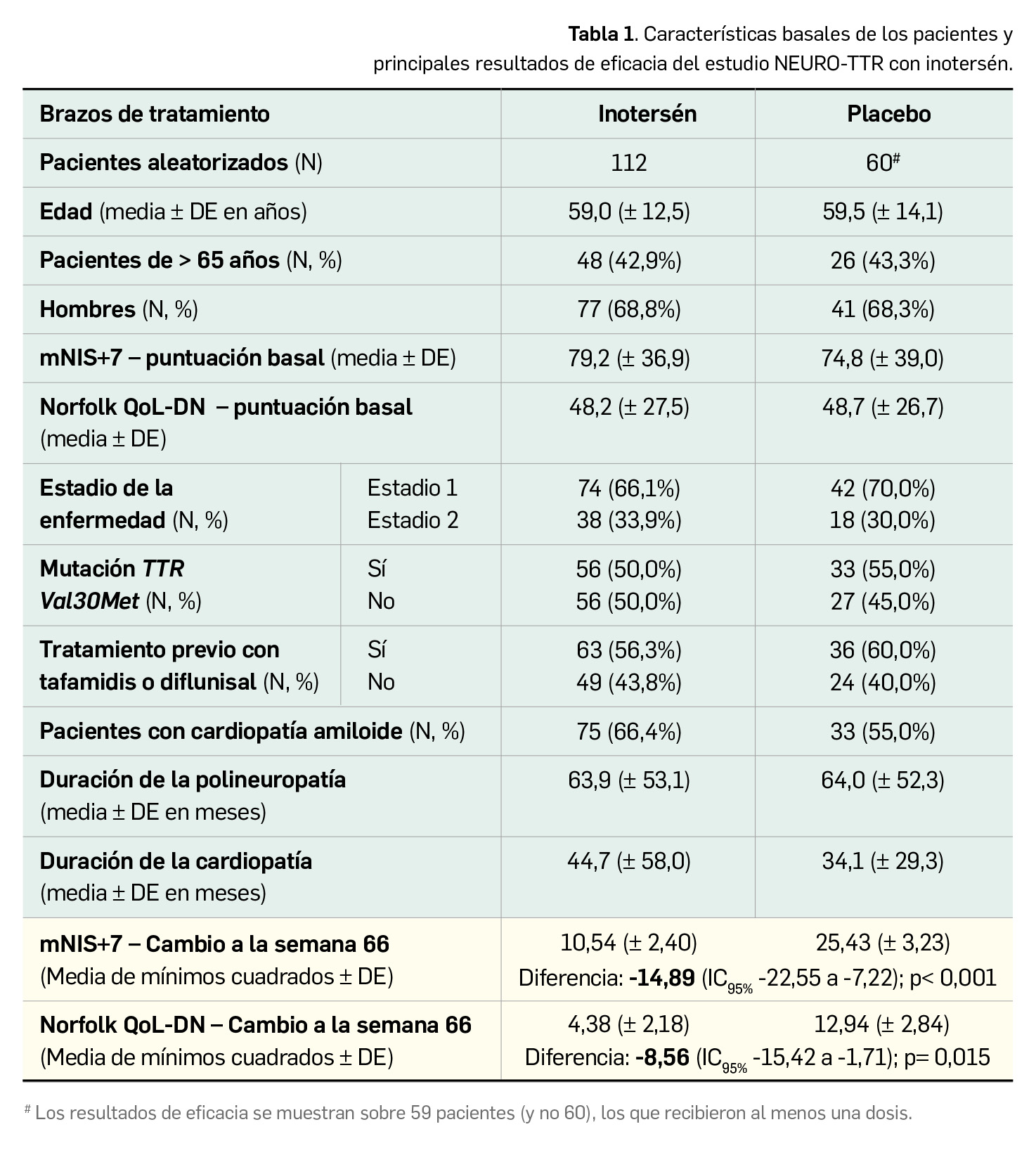{kind=link}
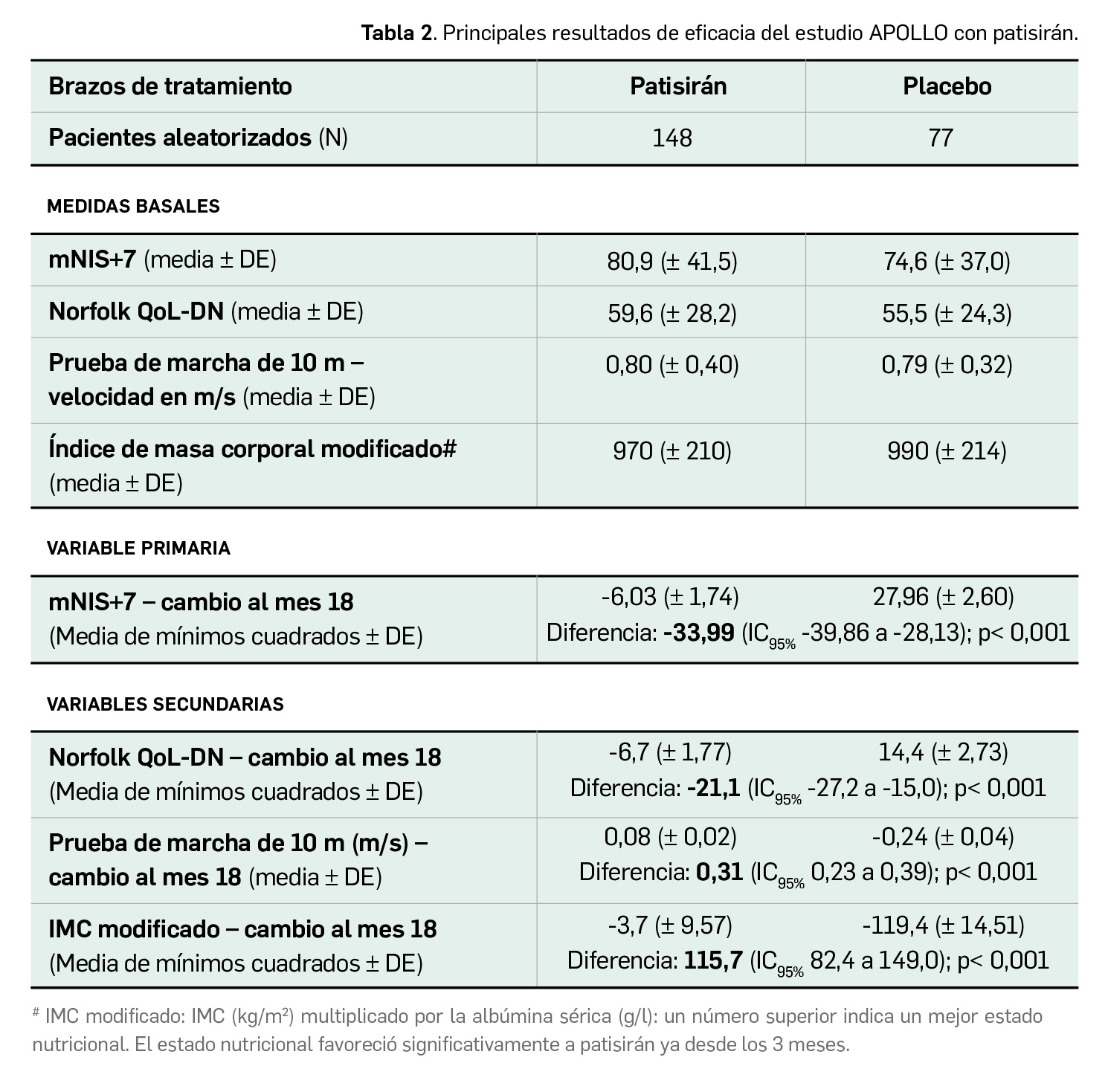{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
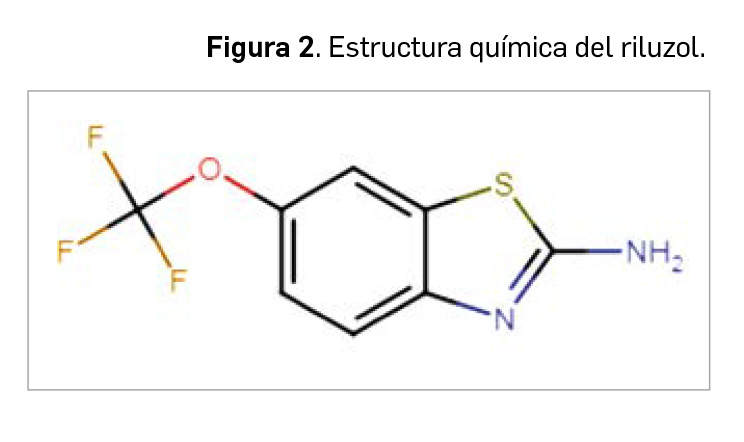{kind=link}
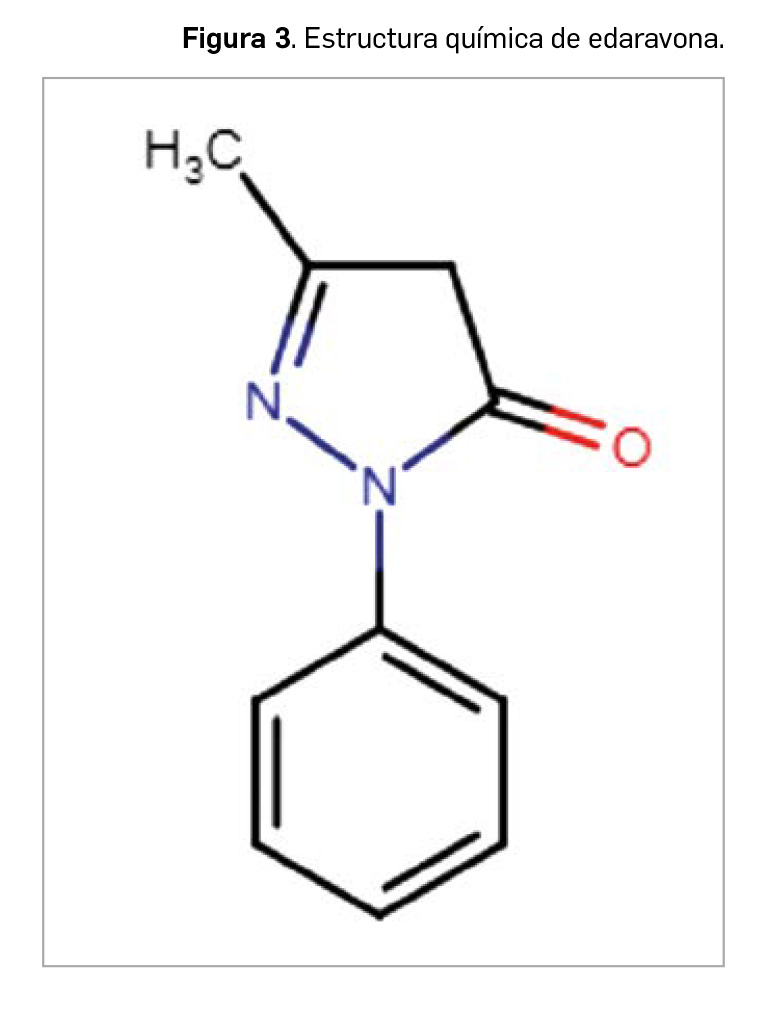{kind=link}
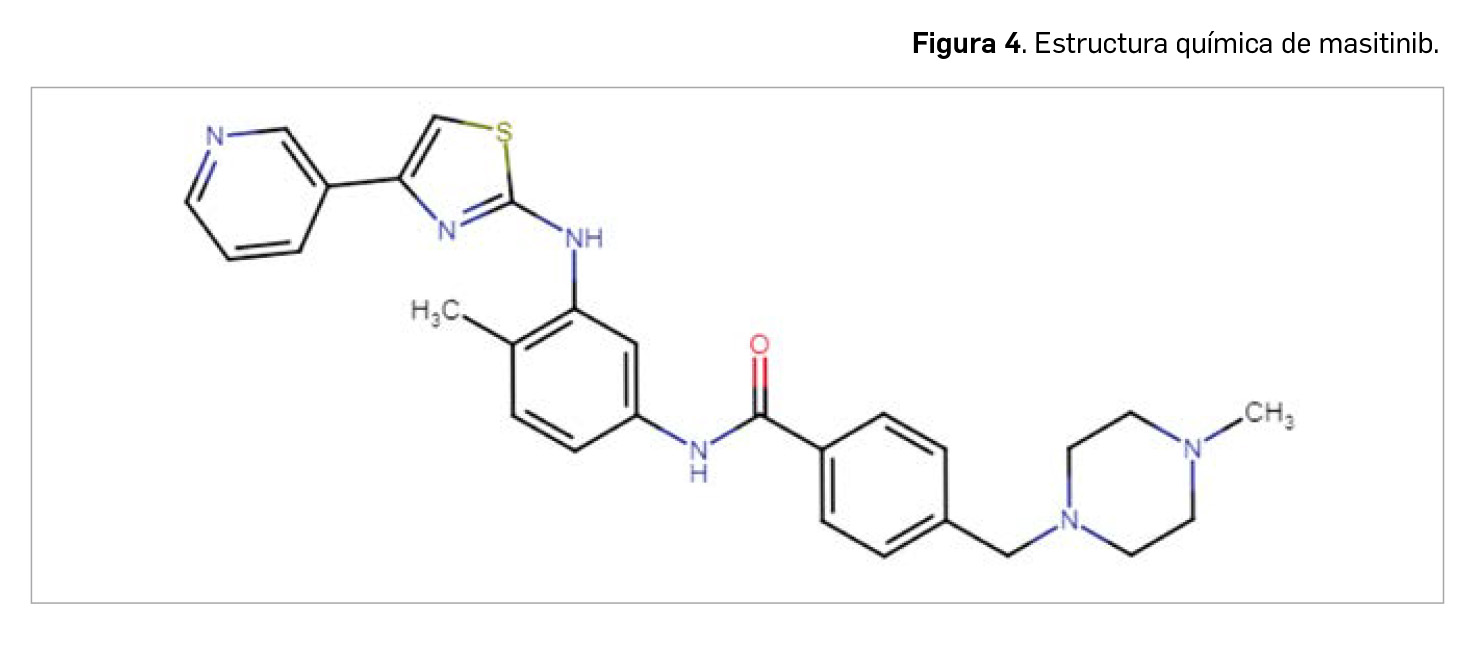{kind=link}
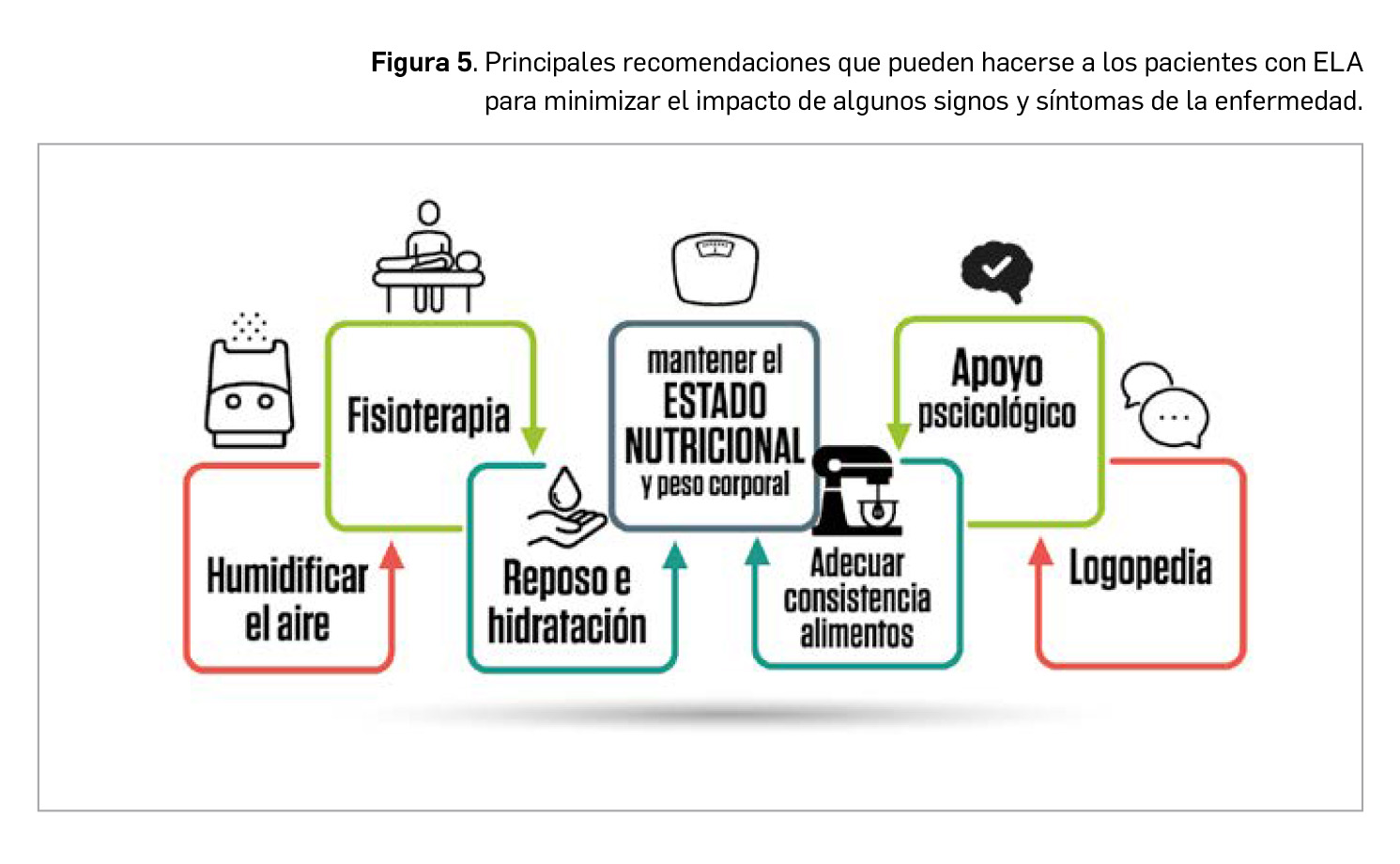{kind=link}
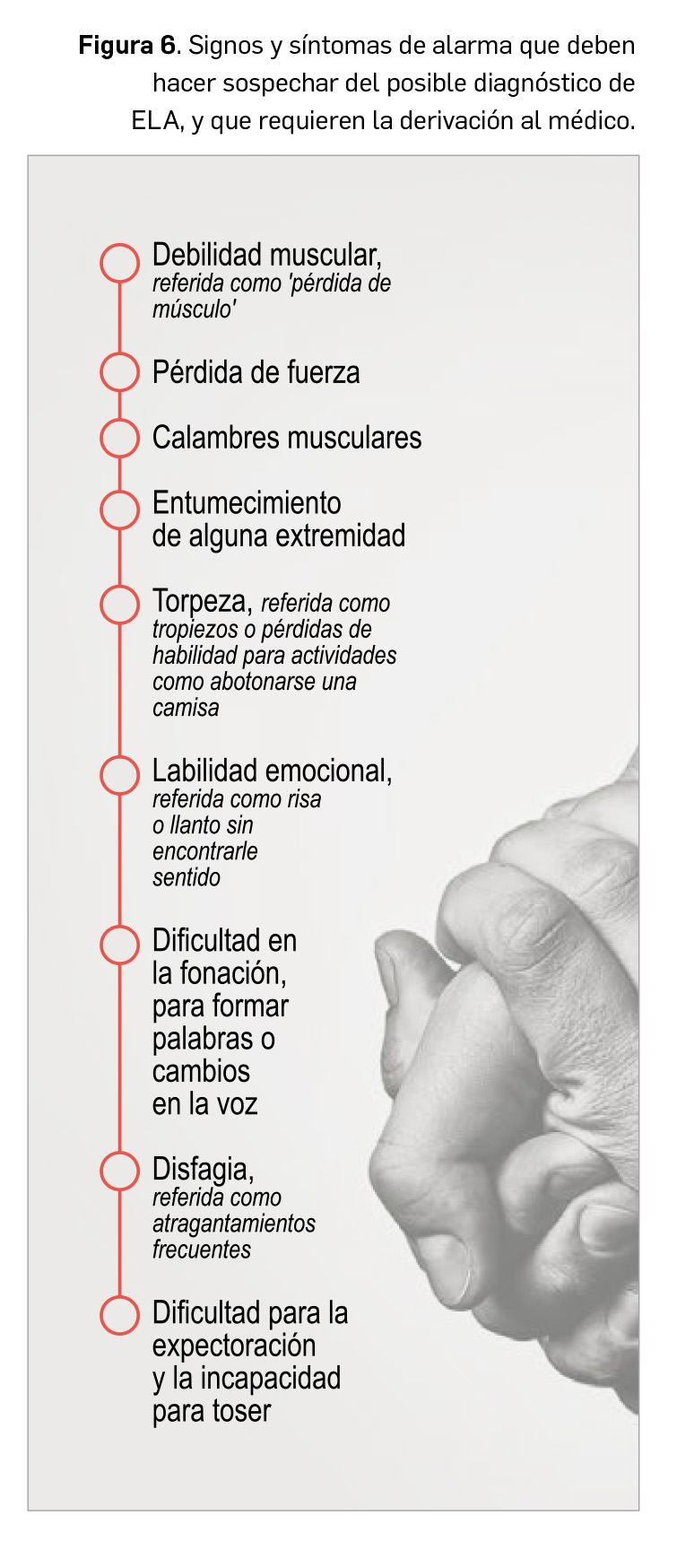{kind=link}